Article Text

Abstract
In the pituitary gland, activating mutations of the GNAS1 (Gsα) gene at Gln227 have been identified in adrenocorticotrophin secreting, growth hormone secreting, and prolactin secreting adenomas. To date, mutations at the codon encoding R201, typically underlying the McCune-Albright syndrome and isolated fibrous dysplasia of bone, have been demonstrated only in growth hormone secreting pituitary adenomas. In this study, a polymerase chain reaction amplified target sequence in exon 8 of the GNAS1 gene was sequenced, identifying the first R201 mutation seen in an isolated basophilic adenoma which generated Cushing's disease in a child. This case adds Cushing's disease to the range of human diseases caused by R201 mutations of the GNAS1 gene.
- ACTH, adrenocorticotrophin
- MAS, McCune-Albright syndrome
- PCR, polymerase chain reaction
Statistics from Altmetric.com
Activating missense mutations of the GNAS1 gene, encoding the α subunit of the stimulatory G protein, Gs (OMIM *139320), occur at codons specifying R201 and Q227 and underlie different clinical disorders. These mutations result in amino acid substitutions that severely impair the intrinsic GTPase activity of the Gsα protein, resulting in the constitutive activation of the Gsα–cAMP signalling pathway in mutated cells.1 Activating mutations of the GNAS1 gene are associated with certain endocrine tumours, hence the denomination of gsp oncogenes, a term that is sometimes used for the mutated alleles.2 Mutations within the codon specifying R201 (R201C or R201H, and more rarely other amino acid substitutions3), are typically associated with the McCune-Albright syndrome (MAS), a congenital, non-inherited disease identified by the triad of polyostotic fibrous dysplasia, skin hyperpigmentation, and different endocrine dysfunctions4. Non-MAS endocrine tumours and tumour-like disorders that have been shown to contain the gsp oncogenes include thyroid adenomas and carcinomas, adrenal adenomas and nodular hyperplasia, and somatotroph pituitary adenomas (reviewed by Spiegel1). Pituitary adenomas of adrenocorticotroph cells have been associated with GNAS1 mutations at Q227 only rarely,5 whereas R201 mutations have never been seen in these lesions. Here, we report an R201H mutation of the GNAS1 gene in an adrenocorticotrophin (ACTH) secreting pituitary adenoma from a child with isolated, pituitary, non-MAS associated Cushing's disease.
“Activating mutations of the GNAS1 gene are associated with certain endocrine tumours”
CASE HISTORY
The patient was an 11 year old girl seen for progressive weight gain and a decrease in linear growth, first noted at the age of 9. The weight gain was associated with facial plethora, hirsutism, dorsocervical and supraclavicular fat accumulation, purple striae on the breast, and mood disturbances. The clinical suspicion of Cushing's disease was confirmed with endocrine studies, which revealed an increase in the 24 hour urinary cortisol, loss of diurnal variation in the serum cortisol, raised serum ACTH, and lack of suppression of the serum cortisol in response to a 1 mg night time dose of dexamethasone. In addition, she had reduced gonadotrophin secretion, with clinical signs of pubertal delay as a result of hypercortisolism. A magnetic resonance imaging scan of the sellar region identified a 2.5 mm left sided microadenoma. Her past medical history also showed a loss of pigment over large tracks of skin, which had begun at approximately 6 years of age. At the age of 9, this area began to re-accumulate pigment. Transphenoidal resection of the pituitary adenoma was performed at the age of 11 years and 9 months, and histopathology of the resection material demonstrated a basophilic adenoma (fig 1A). Immunohistochemically, the tumour was positive for ACTH and low molecular weight cytokeratins, and negative for growth hormone, prolactin, β thyrotrophin, β luteinising hormone, β follicle stimulating hormone, and the α-subunit of glycoprotein hormones. In the postoperative period, signs and symptoms of Cushing's disease persisted, in addition to increased concentrations of urinary cortisol and plasma ACTH, possibly because of incomplete ablation of the ACTH producing lesion, or cell population. At the age of 12 years and 6 months the patient received stereotactic radiation treatment (45 Gy) of the sellar area. Currently (age 13 years and 11 months), hormonal values are approaching the normal range (urinary cortisol, 59.4 μg/24 hours; serum cortisol at 8.00 AM, 118 μg/litre; plasma ACTH at 8.00 AM, 22.7 pg/ml), and the clinical signs of Cushing's disease are resolving as indicated by weight loss, pubertal progression into menarche, mood improvement, and a lack of clinical or biochemical signs of hypopituitarism.

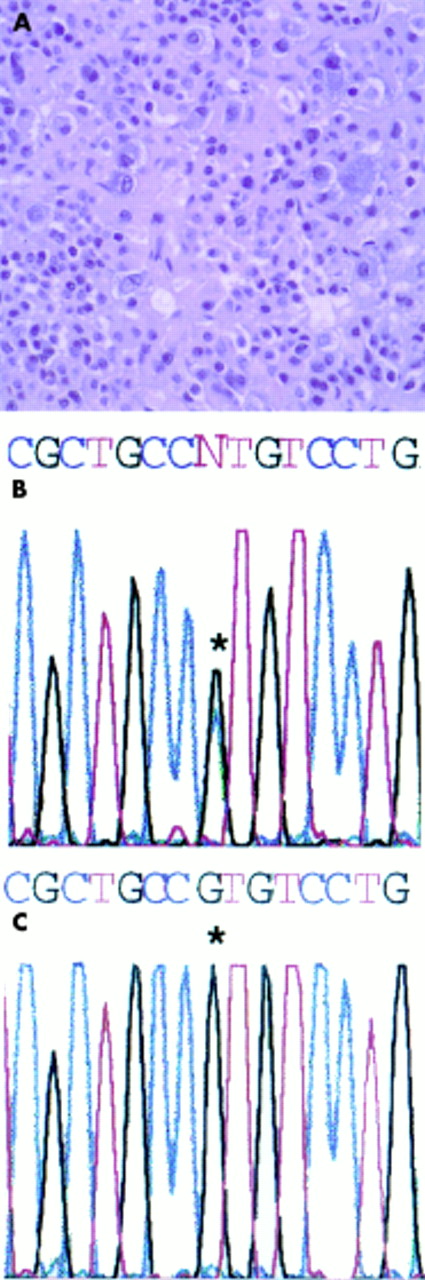{kind=link}
(A) Overview of the histology of the pituitary adrenocorticotrophin producing growth. (B) Sequence of the PCR product obtained by amplifying the relevant target sequence of exon 8 of the GNAS1 gene. A G → A transition at the codon specifying R201 (asterisk) indicates an R201H missense mutation. (C) Normal genotype in the genomic DNA extracted from a skin sample.
MATERIALS AND METHODS
Because of the concurrence of an endocrine disorder with a generically defined disorder of skin pigmentation, ruling out a diagnosis of MAS was felt to be clinically relevant. In retrospect, the type and evolution of the cutaneous pigmentation disorder were inconsistent with “café au lait spots” characteristic of MAS, and rather typical of vitiligo. A sample of the transphenoidal resection material and a skin biopsy were used for mutation analysis. DNA was extracted from fresh pituitary tissue and skin using a commercially available kit according to the manufacturer's instructions (Qiagen, Valencia, California, USA). A 300 bp fragment of exon 8 of the Gsα gene, containing the codon for R201, was amplified in a standard polymerase chain reaction (PCR) using the following primers (GenBank Accession number M21142): 5`-TGACTATGTGCCGAGCGA (forward, exon 7, bases 420–438) and 5`-AACCATGATCTCTGTTATATAA (reverse, intron G, bases 728–746).
After purification by the Wizard PCR PREP DNA purification system (Promega, Madison, Wisconsin, USA), the amplification product was sequenced by the dideoxy chain termination method using an ABI 370 automated DNA sequencer and the ABI PRISM dye terminator cycle sequencing ready reaction kit (Perkin Elmer, Norwalk, Connecticut, USA).
RESULTS
DNA sequencing of the PCR amplification product of the pituitary genomic DNA showed a G → A replacement in the codon for R201, indicating an R201H mutation (fig 1B). No mutation was detected in the R201 codon in DNA extracted from the skin biopsy (fig 1C), indicating that the hyperpigmentation was not related to a Gsα mutation in the skin, at variance with MAS.
Take home message
-
These results add Cushing's disease to the range of human diseases for which R201 somatic mutations of the GNAS1 gene may be responsible
DISCUSSION
The results of our report add isolated pituitary Cushing's disease to the range of human diseases for which R201 somatic mutations of the GNAS1 gene may be responsible. Activating missense mutations at the codon specifying R201 are typical of isolated, monostotic or polyostotic fibrous dysplasia of bone6 and of MAS4, in which skeletal fibrous dysplasia (resulting from mutation of osteogenic cells7), endocrine disorders, and skin pigmentation concur. GNAS1 mutations found in isolated endocrine tumours include mutations at R201 and Q227.2 These mutations are thought to be relatively common in somatotroph adenomas, although their estimated frequency varies greatly (9–42%) in different reported series.2,8,9 In contrast, they are believed to be associated with corticotroph adenomas only rarely.5 All cases of Cushing's disease occurring in the context of MAS appear to represent ACTH independent adrenal disease (adenoma or nodular hyperplasia) (reviewed in Collins and colleagues10). To date, only two cases of ACTH producing adenomas have been recorded in which a gsp allele was demonstrated, and in both cases the mutation was at Q227, not at R201.5 However, the two instances of Q227 mutations previously reported still represent 6% (two of 32) of the only sizable series of corticotroph adenomas investigated to date. Hence, in light of our finding of an R201 mutation, the real frequency of GNAS1 mutations in pituitary Cushing's disease remains to be determined and may well be in the same range as for somatotroph adenomas.
Acknowledgments
The support of Telethon Fondazione Onlus (E1029) to PB is gratefully acknowledged.