Article Text

Abstract
Background and aims: Although it is reported that Helicobacter pylori induces apoptosis on gastric epithelial cells, the mechanism remains unknown. Antiapoptotic effects generated by H pylori have not yet been evaluated.
Methods: (1) H pylori strains (type 1 wild, TN2-ΔcagE, TN2-ΔvacA) were cocultured with MKN45, TMK1, and HeLa cells, and cell viability and apoptosis were assessed by trypan blue exclusion and DNA laddering, respectively. (2) Activation of caspases-3, 7, and 8, cytochrome c release from the mitochondria, and Fas, Fas associated death domain protein (FADD), Bax, Bak, and Bcl-X expression were evaluated by immunoblot analysis. (3) To investigate whether nuclear factor kappa B (NFκB) activation induced by cag pathogenicity island (PAI) positive H pylori affects antiapoptosis, MKN45 cells stably expressing super-repressor Iκβα were cocultured with H pylori, and cell viability and caspase activation were evaluated. NFκB regulated gene expression was also evaluated by ribonuclease protection assay.
Results: (1) Wild-type and ΔvacA mutant H pylori induced apoptosis more potently than the ΔcagE mutant. Inhibition of cell contact between H pylori and cancer cells and heat killing H pylori diminished cell death. (2) Caspases-3, 7, and 8 were activated time dependently by H pylori as well as by the agonist anti-Fas. Cytochrome c release from mitochondria was observed and was not inhibited by caspase-8 inhibitor. Although protein expression of Fas, FADD, Bax, Bak, and Bcl-X in the whole cell lysates was not changed by H pylori, Bax was decreased from mitochondria free cytosol suggesting that Bax was translocated into mitochondria. (3) Cell death and the activities of caspases-3 and 8 were promoted in MKN45 cells stably expressing super-repressor Iκβα that inhibits NFκB activation. Antiapoptotic proteins c-IAP1 and c-IAP2 were upregulated by the wild-type strains.
Conclusion:cag PAI positive H pylori is capable of inducing apoptotic effects mainly through the mitochondrial pathway. Antiapoptotic effects mediated by NFκB activation were also observed.
- Helicobacter pylori
- apoptosis
- antiapoptosis
- signalling pathway
- PAI, pathogenicity island
- NFκB, nuclear factor kappa B
- FADD, Fas associated death domain protein
- IFN, interferon
- PBS, phosphate buffered saline
- BSA, bovine serum albumin
- VacA, vacuolating cytotoxin
- FBS, fetal bovine serum
- MyD88, myeloid differentiation factor 88
Statistics from Altmetric.com
- PAI, pathogenicity island
- NFκB, nuclear factor kappa B
- FADD, Fas associated death domain protein
- IFN, interferon
- PBS, phosphate buffered saline
- BSA, bovine serum albumin
- VacA, vacuolating cytotoxin
- FBS, fetal bovine serum
- MyD88, myeloid differentiation factor 88
Helicobacter pylori is a gram negative bacterium that infects the human stomach1 and plays an important role in the pathogenesis of chronic gastritis and peptic ulcer diseases.2,3 In addition, epidemiological studies have consistently identified an association between H pylori infection and the development of gastric adenocarcinoma and mucosa associated lymphoid tissue lymphoma.4–6 However, the mechanisms underlying the carcinogenic potential of H pylori are not completely understood.
Homeostasis of the gastrointestinal mucosa is maintained through a balance between the proliferation and apoptosis of mucosal cells. Apoptosis is also implicated in carcinogenesis, autoimmune diseases, and various infectious diseases. Although infection with H pylori is associated with significant epithelial cell damage, including an increased level of apoptosis, the mechanism underlying H pylori induced apoptosis in gastric epithelial cells remains unclear.7–10
Two major pathways leading to apoptosis have been described. One pathway involves apoptosis mediated by death receptors, such as CD95 (Fas) and tumour necrosis factor receptors. When the Fas ligand binds to the Fas receptor, formation of the death inducing signal complex comprising the adapter molecule Fas associated death domain protein (FADD) and caspase-8 results in the active caspase-8 and process effector caspases (caspases-3, 6, and 7), thereby inducing apoptosis.11,12 In the other pathway, various proapoptotic signals converge at the mitochondria level, provoking translocation of cytochrome c from the mitochondria to the cytoplasm. Once cytochrome c is released into cytoplasm, it binds to Apaf-1 and induces recruitment of procaspase-9. Activated caspase-9 then cleaves and activates procaspase-3. Bcl-2 family members are associated with mitochondria related apoptosis. While cell survival-promoting molecules Bcl-2 and Bcl-X, localised at the outer mitochondrial membrane, prevent translocation of cytochrome c from the mitochondria, induced expression or enforced dimerisation of Bax results in mitochondrial dysfunction leading to cytochrome c release.13–15
Several studies reported that the Fas/Fas ligand system was involved in H pylori induced apoptosis.10,16,17 In these reports, H pylori strains or supernatant upregulated Fas/Fas ligand expression and induced apoptosis indirectly. However, it is not known if these systems are major pathways of H pylori mediated apoptosis. Moreover, the other main apoptotic pathway, the mitochondrial pathway, was not investigated. In contrast, there are a few reports of an association between the Bcl-2 family, which is involved in the mitochondrial pathway, and H pylori induced apoptosis, where upregulation of Bak or Bax was associated with H pylori induced apoptosis in vitro or in vivo.18,19 However, these studies did not investigate most of the other proteins associated with the apoptotic pathway.
Several factors have been proposed as possible virulence determinants of H pylori. In particular, cag pathogenicity island (cag PAI), a 40 kb region of possibly extraneous origin, is responsible for transcriptional factor nuclear factor kappa B (NFκB) activation.20–22 Isogenic mutant studies demonstrated that some proteins encoded by cag PAI genes are responsible for NFκB activation.23 NFκB is a regulator of genes involved in inflammation, cell proliferation, and apoptosis.24,25 Recent studies suggest that NFκB may play a critical role in protecting cells against apoptosis.26,27 The antiapoptotic role played by NFκB involves the ability of this transcriptional factor to induce expression of genes that promote cell survival such as the genes coding for TRAF1, TRAF2, and the cellular inhibitors of apoptosis 1 and 2 (c-IAP1, c-IAP2).28 Curiously, NFκB has been found to be associated with proapoptotic as well as antiapoptotic mechanisms. For instance, NFκB activation appears to induce apoptosis in cells exposed to hydrogen peroxide.29 The magnitude of the stimulus and the cell type involved may determine whether NFκB leads to cell survival or cell death.
Although H pylori infection induces apoptosis in gastric epithelial cells, the mechanism of intracellular signal conduction that leads to apoptosis is scarcely known. In addition, it is not known whether H pylori mediated NFκB activation plays an apoptotic or antiapoptotic role. The aims of this study were to clarify the molecular mechanism of the proapoptotic pathway induced by H pylori, and to investigate the relation between H pylori induced NFκB activation and apoptosis.
MATERIALS AND METHODS
Bacterial strains
TN2, a strain positive for CagA, cag PAI, and VacA (vacuolating cytotoxin), were generously provided by Dr Nakao (Takeda Chemical Industries Ltd, Osaka, Japan). Infection with this strain induces gastric cancer in Mongolian gerbils.30 Isogenic cagE negative and vacA negative mutants, TN2-ΔcagE, and TN2-ΔvacA were prepared by insertion of a kanamycin resistant gene into the cagE and vacA locus of TN2, as previously described.31,32H pylori strains were cultured on Columbia agar with 5% (vol/vol) horse blood and Dent antibiotic supplement (Oxoid, Basingstoke, UK) at 37°C for three days under microaerobic conditions (Campy-Pak Systems; BBL, Cockeysville, Maryland, USA). The isolates were kept at −80°C in Brucella broth with 5% (vol/vol) fetal bovine serum (FBS) containing 16% (vol/vol) glycerol. In coculture experiments, H pylori was cultured in Brucella broth containing 7.5% FBS for 24 hours, centrifuged, and resuspended in cell culture medium (RPMI 1640) containing 10% FBS, and then applied immediately to assays. The range of bacteria:cancer cell ratio was 50:1 to 75:1 when cocultured with 107 colony forming units/ml. Heat killed H pylori bodies were prepared by heating at 80°C for 30 minutes.
Plasmids and reagents
The super-repressor mutant of IκBα, IκBα (SS32/36AA) subcloned in pcDNA3, was generously donated by Dr Suzuki (Yamanouchi Pharmaceutical Co, Ltd, Ibaraki, Japan).33 The anti-FLAG monoclonal antibody M2 antibody was purchased from Sigma (St Louis, Missouri, USA); the polyclonal anti-Fas antibody, polyclonal anti-IκBα, and anti-Bak antibody from Santa Cruz Biotechnology (Santa Cruz, California, USA); the monoclonal anti-actin antibody from Chemicon International (Temecula, California, USA); the monoclonal agonist anti-Fas antibody CH-11, monoclonal neutralising anti-Fas antibody ZB4, monoclonal anti-caspase-3, monoclonal anti-caspase-7, and monoclonal anti-caspase-8 from MBL (Nagoya, Japan); polyclonal cleaved caspase-3 from Cell Signaling Technology (Beverly, Massachusetts, USA); the monoclonal anti-FADD antibody, monoclonal anti-Bcl-X monoclonal, and anti-Bax antibody from Transduction Laboratories (San Diego, California, USA); the polyclonal anti-cytochrome c antibody from Pharmingen (San Diego, California, USA); Ac-IETD-CHO from Peptide Institute, Inc (Osaka, Japan); and recombinant human interferon γ (IFN-γ) from Pepro Tech EC (London, UK).
Human cell lines
Human gastric cancer cells MKN45 and TMK1 were maintained in RPMI-1640 containing 10% FBS, l-glutamine, 100 U of penicillin-G, and 100 μg/ml of streptomycin. MKN45 was obtained from the Riken Gene Bank (Tsukuba, Japan). TMK1 was provided by Dr E Tahara (Hiroshima University, Hiroshima, Japan). HeLa cells were maintained in Dulbecco's modified Eagle's medium containing 10% FBS, l-glutamine, 100 U of penicillin-G, and 100 μg/ml of streptomycin. HeLa cells were obtained from the Riken Gene Bank.
Cell viability assay
Cells were collected and washed in phosphate buffered saline (PBS). Cell viability was assessed by trypan blue dye exclusion assay by counting 300 cells. Viable cells were quantitated under bright field microscopy.
Analysis of DNA fragmentation
Cancer cells cocultured with H pylori or treated with anti-Fas (CH-11) (approximately 1×105cells) were washed in PBS and lysed in 40 μl of lysis buffer containing 200 mM Na2HPO4 and 4 mM citric acid. Samples were centrifuged and the supernatants were incubated with 3 μl of 0.25% NP-40 and 3 μl of Dnase-free Rnase (10 mg/ml), and followed with a 10 mg/ml proteinase K digestion. Aliquots (10 μl) from a 50 μl DNA solution were electrophoresed on a 2% agarose gel.
Immunoblot analysis
Cells were suspended in 50 mM Tris HCl (pH 7.4) buffer containing 1 mM EGTA, 2 mM DTT, 25 mM sodium β-glycerophosphate, 0.1 mM PMSF, and 10 μg/ml aprotinin. An equal amount of protein extracts was fractionated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis and electrophoretically transferred to a PVDF membrane (Amersham Pharmacia Biotech, Buckinghamshire, UK). The membrane was probed with the antibodies described above. An ECL detection assay (Amersham Pharmacia Biotech) was performed according to the manufacturer's instructions.
Immunoprecipitation
MKN45 cells (5×106) were incubated with H pylori or the agonist anti-Fas for 12 hours, and lysed in 50 mM Tris-HCl (pH 7.4) buffer containing 1 mM EGTA, 2 mM DTT, 25 mM sodium β-glycerophosphate, 0.1 mM PMSF, and 10 μg/ml aprotinin. Lysates were immunoprecipitated with anti-Fas antibody (Santa Cruz) and 20 μg of protein A-Sepharose. Immunoprecipitates were washed five times in PBS and used for immunoblot analysis.
Generation of stable transfectants
MKN45 (5×105) was seeded onto 10 cm plates and transfected 24 hours later with 3 μg of FLAG-IκBα (SS32/36AA) subcloned in pcDNA3 or control vector pcDNA3 using Effectene transfection reagent (Quiagen, Hilden, Germany). Cells stably expressing FLAG-IκBα (SS32/36AA) were selected in medium containing 1 mg/ml G418 (Gibco BRL, Life Technologies, Inc., Rockville, Maryland, USA) for two weeks. MKN45 cells transfected with the pcDNA3 vector and selected by G418 were used as controls.
RNA isolation and nuclease protection assay
Total RNA was extracted using an acid guanidinium thiocyanate-phenol-chloroform method according to the manufacturer's instructions (Isogen; Nippongene, Tokyo, Japan). Total RNA (10 μg) was hybridised with antisense RNA probes labelled with 32P-UTP using the apoptosis related template set, hAPO-2 (bcl-X L/S, bfl1, bik, bax, bcl-2, mcl-1, and internal controls of L32 and GAPDH) or hAPO-5 (XIAP, TRAF1, TRAF2, TRAF4, NAIP, c-IAP-2, c-IAP-1, TRPM2, TRAF3, and internal controls of L-32 and GAPDH), purchased from Pharmingen. The RNA duplexes were run on 5% polyacrylamide gels and autoradiographically scanned using a FLA3000 image analyser (Fuji Photo Film Co, Ltd, Tokyo, Japan).
DNA fragmentation ELISA
DNA fragmentation was quantified using a commercially available ELISA (Boehringer Mannheim Biochemicals, Mannheim, Germany) that detects nucleosomal fragments in the cytoplasmic fractions of cells undergoing apoptosis but not necrosis. For these experiments, 5×104 cells were incubated in triplicate with H pylori, anti-Fas (CH-11), or medium alone for 12 hours and lysed, and supernatants were used for ELISA. Absorbance was measured at 405 nm.
Electrophoretic mobility shift assay
Detection of NFκB was performed with a 32P dATP labelled oligo probe containing the NFκB recognition site purchased from Promega (Madison, Wisconsin, USA). The DNA binding reactions were performed at room temperature for 30 minutes in a 10 μl mixture consisting of 4% glycerol, 1 mM MgCl2, 0.5 mM EDTA, 0.5 mM dithiothreitol, 50 mM NaCl, and 0.5 μg of poly(dI-dC). Supershift analysis was performed using antibodies against p65 and p50. DNA-protein complexes were loaded onto a chilled 4% non-denaturing acrylamide gel. Gel electrophoresis was executed in 0.5×Tris borate-EDTA at 4°C. The gel was dried and autoradiography was performed using a Fujix bioimaging analyser FLA 3000 (Fuji Photo Film).
Statistics
Data are presented as means (SEM). Differences in means were examined by ANOVA with ad hoc test. A p value <0.05 was considered significant.
RESULTS
H pylori induced apoptosis in cancer cells
Apoptosis induction was evaluated in vitro in three cancer cell lines, MKN45, TMK1, and HeLa. Cells were treated with or without IFN-γ (10 ng/ml) for 24 hours and then incubated with H pylori (TN2) (bacteria:cancer cell ratio 50:1–75:1) or the agonist anti-Fas (CH-11). DNA fragmentation was induced in all cell lines 16 hours after coculture with H pylori as well as after anti-Fas treatment with IFN-γ (data not shown). Weak DNA fragmentation was observed both after coculture with H pylori and after treatment with anti-Fas without IFN-γ (data not shown). We tested other bacterial ratios, 5:1 and 500:1. At 5:1, neither apparent cell death nor apoptosis was observed. At 500:1, cells lost viability immediately after addition of bacteria, with or without IFN-γ priming, making it impossible to analyse apoptosis. A bacteria:cancer cell ratio of 50:1–75 was used in the following studies. Exposure of MKN45, TMK-1, and HeLa (with or without IFN-γ) cells to H pylori or anti-Fas caused a time dependent decrease in the number of viable cells, as determined by the trypan blue dye exclusion assay (fig 1). These results indicate that H pylori directly induces cell apoptosis. Pretreatment with IFN-γ strengthened the apoptotic effects by H pylori and the agonist anti-Fas in these cell lines. Pretreatment with IFN-γ was performed in the following studies.

Incubation with Helicobacter pylori (HP) cells decreased the viability of cancer cells in the three cancer cell lines. Cell viability was assessed by trypan blue dye exclusion assay at the indicated times by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of viable cells. Values are mean (SD) of three independent experiments. *Percentage viability was significantly (p<0.05) different between the control group at 36 hours and the H pylori coculture or anti-Fas treatment without interferon γ (IFN-γ) group. †Percentage viability was significantly (p<0.05) different between the H pylori coculture or anti-Fas without IFN-γ group and the IFN-γ group.
Effects of H pylori virulence factors
To evaluate the effects of H pylori virulence factors, the cagE mutant TN2-ΔcagE and the vacA mutant TN2-ΔvacA were used. In the cell viability assay, wild-type and TN2-ΔvacA significantly decreased the viability of MKN45 and TMK-1 cells after 24 and 36 hours of coculture (fig 2A). The DNA fragmentation assay revealed that wild-type and TN2-ΔvacA induced apoptosis after 16 hours of infection whereas TN2-ΔcagE did not induce apoptosis at this time (fig 2B). However, cagE also induced apoptosis after 36 hours of infection (data not shown). These results indicate that the cagE mutant induces apoptosis less effectively than the wild-type or vacA mutant.
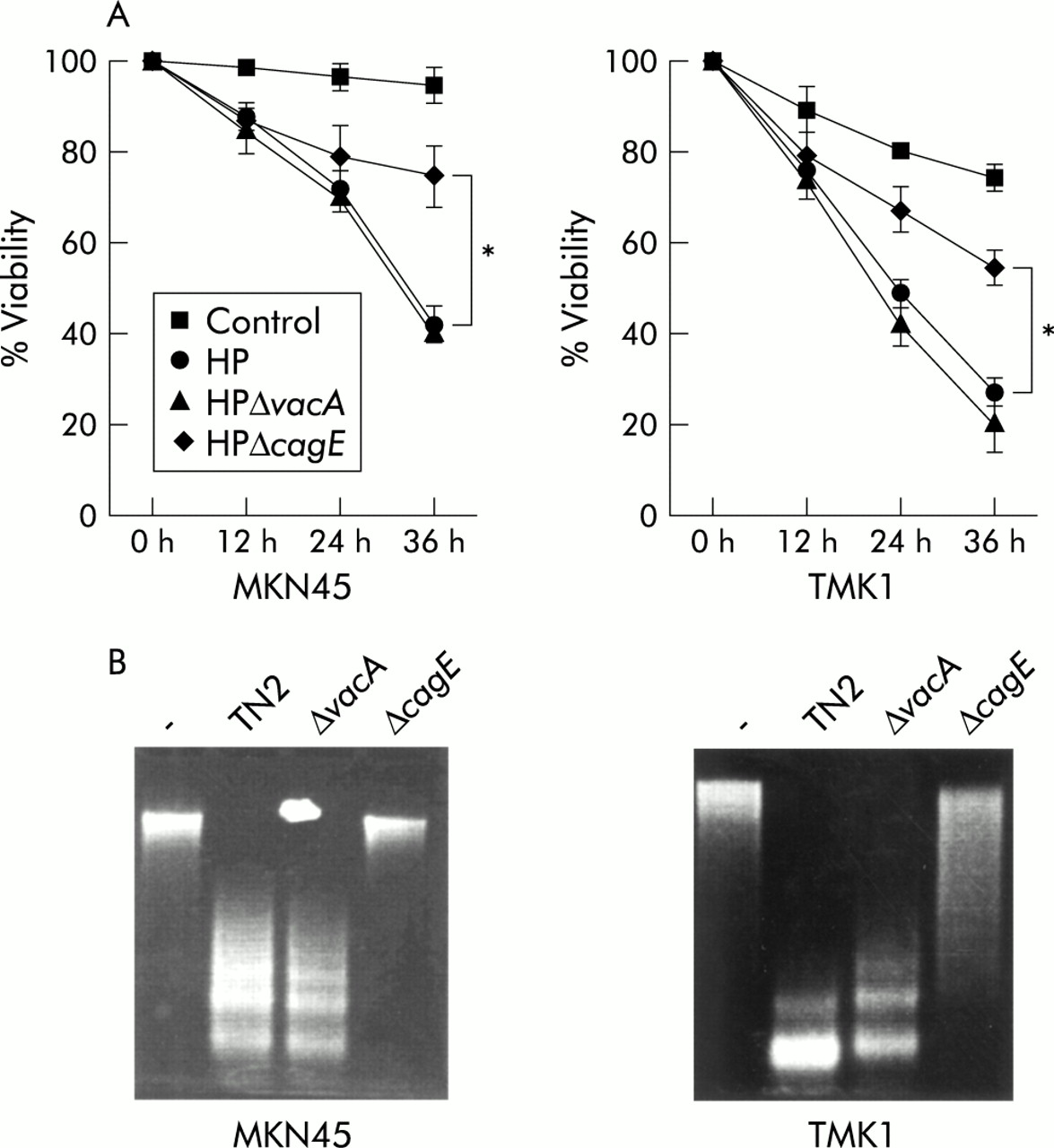
Attenuation of the apoptotic effect by cagE negative mutant. (A) Cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours and incubated with H pylori (TN2), TN2-ΔcagE, and TN2-ΔvacA. Cell viability was assessed by trypan blue dye exclusion assay at the indicated times by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of viable cells. Values are mean (SD) of three independent experiments. *Percentage viability was significantly (p<0.05) different between the H pylori (HP) wild-type and vacA coculture group and the cagE coculture group. (B) DNA fragmentation was evaluated after 16 hours of infection.
Effects of heat killed H pylori and inhibition of direct contact with host cells
To assess the effect of direct contact, cancer cells and bacteria were separated by a membrane filter (Nunc Tissue Culture Inserts No 162138; Nunc, Roskilde, Denmark). We also used heat killed bacteria heated at 80°C for 30 minutes. These procedures suppressed the H pylori mediated decrease in cell viability (fig 3). No DNA fragmentation was observed with heat killed bacteria or with viable bacteria separated by a permeable membrane, indicating that direct contact with viable bacteria is necessary for induction of apoptosis by H pylori.

Viable bacteria and direct contact with cells were necessary for inducing apoptosis. MKN45 cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours. Cells and bacteria were separated by membrane filter (Nunc Tissue Culture Inserts No 162138; Nunc, Roskilde, Denmark). We also used heat killed bacteria at 80°C for 30 minutes. Cell viability was assessed by trypan blue dye exclusion assay at 24 hours by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of dead cells. Values are mean (SD) of three independent experiments. *Percentage cytotoxicity was significantly (p<0.05) different between the H pylori (HP) coculture group and the heat killed and separated by filter groups.
Activation of caspases-8, 3, and 7
To assess whether H pylori induces apoptosis via caspase activation, immunoblot analysis was performed using anti-caspases-8, 3, 7, and cleaved caspase-3 antibodies in MKN45 cells. In untreated MKN45 cells, caspase-8 was present primarily as a molecule of approximately 55 kDa. Incubation with H pylori or the agonist anti-Fas resulted in a time dependent degradation of the primary form and processing into two fragment of 43 and 41 kDa intermediate forms. Caspase-8 processing increased eight hours after H pylori treatment. In untreated MKN45 cells, caspase-3 was present primarily as a molecule of approximately 32 kDa. Incubation with H pylori or anti-Fas resulted in a time dependent degradation of the primary form that was processed to a 20 kDa active form. The active form of caspase-3 was observed 12 hours after H pylori treatment. In untreated MKN45 cells, caspase-7 was present primarily as a molecule of approximately 35 kDa. Incubation with H pylori and anti-Fas resulted in a time dependent degradation of the primary form. An increase in the processing of caspase-7 was observed 12 hours after H pylori treatment. These results indicate that H pylori and anti-Fas activated caspases-8, 3, and 7 in MKN 45 cells (fig 4). Similar results were obtained using TMK-1 cells (data not shown).
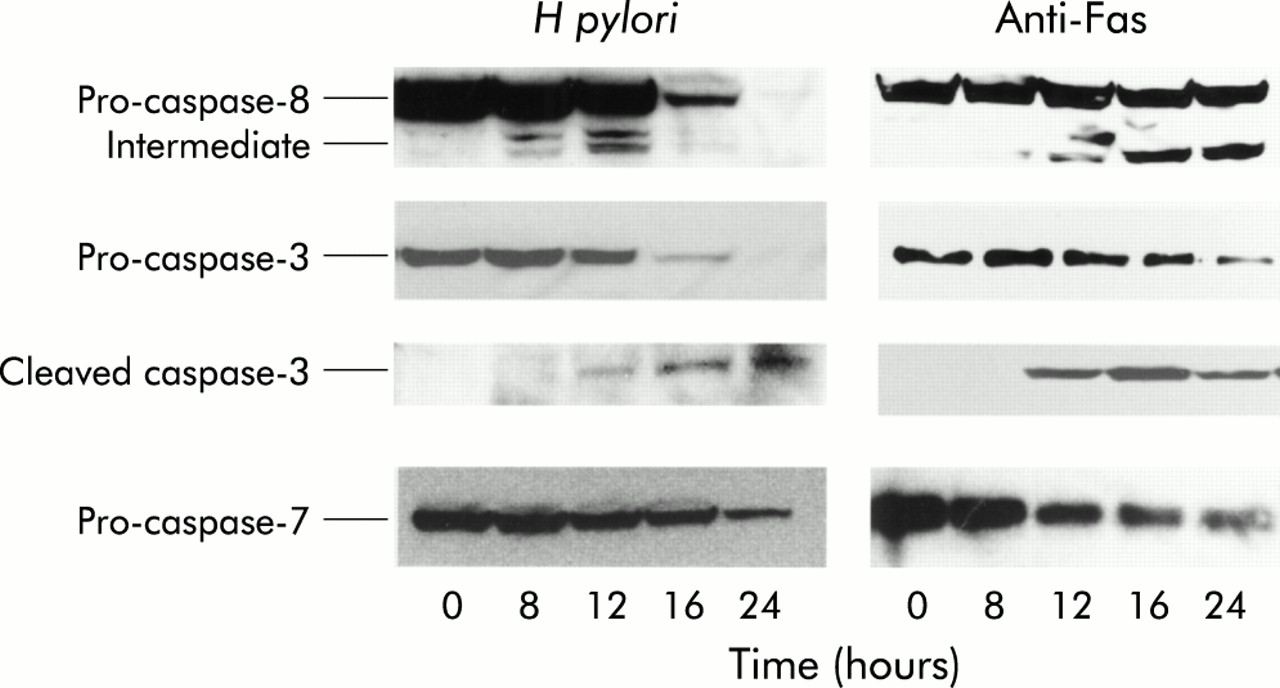
Helicobacter pylori induces apoptosis via a caspase dependent pathway. Immunoblot analysis was performed using anti-caspases-8, 3, 7, and cleaved caspase-3 antibodies in MKN45 cells. Incubation with H pylori and anti-Fas resulted in a time dependent degradation of the primary forms of caspases-8, 3, and 7. Processing into two fragments (43 kDa and 41 kDa) of the caspase-8 intermediate form and the 20 kDa active caspase-3 was also observed.
Caspase-8 and Fas do not play a major role in H pylori induced apoptosis
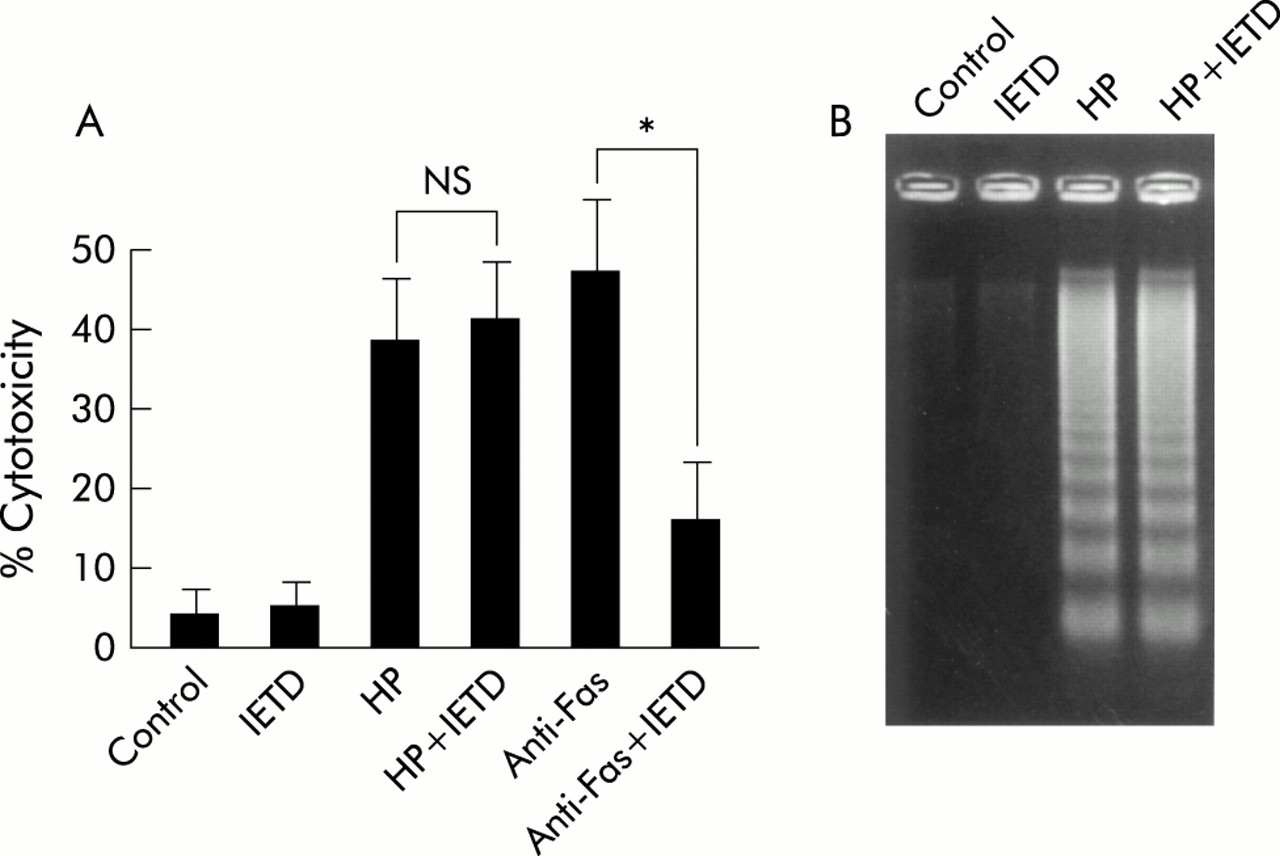
We evaluated the effect of the caspase-8 inhibitor Ac-IETD-CHO on H pylori mediated apoptosis in MKN45 cells. While the inhibitor (100 μM) abolished anti-Fas induced cell death (from 50% to 15%), H pylori mediated cell death was not affected (fig 5A), and DNA fragmentation induced by H pylori was not inhibited (fig 5B). These results indicate that caspase-8 activation by H pylori does not function as a major pathway in H pylori induced apoptosis. Similar results were obtained using TMK-1 cells (data not shown).

Caspase-8 inhibitor did not inhibit Helicobacter pylori induced apoptosis. (A) MKN45 cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours. Cells were preincubated with or without the caspase-8 inhibitor Ac-IETD-CHO (IETD 100 μM) for one hour and then incubated with H pylori (HP) or anti-Fas (CH-11) for 24 hours. Cell viability was assessed by trypan blue dye exclusion assay by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of dead cells. Values are mean (SD) of three independent experiments. *Percentage cytotoxicity was significantly (p<0.05) different between the anti-Fas treatment group and the anti-Fas treatment with caspase-8 inhibitor treatment group. NS, no significant difference was found. (B) DNA fragmentation was also evaluated after 24 hours of infection.
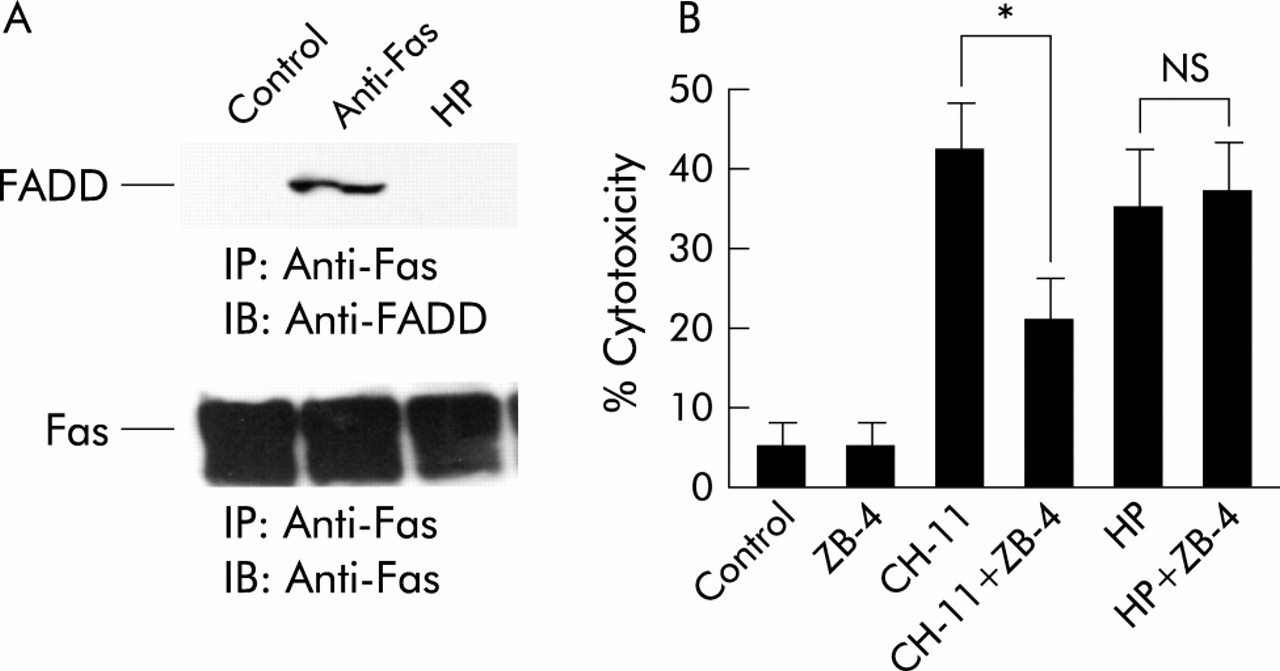
We evaluated whether FADD was recruited to Fas and found that under apoptosis induced conditions, anti-Fas but not H pylori recruited FADD to Fas (fig 6A). In evaluating the effect of neutralising anti-Fas antibody (ZB-4), we found that ZB-4 decreased cell cytotoxicity induced by the agonist anti-Fas antibody (CH-11) (from 42% to 21%) but not H pylori mediated cell cytotoxicity (from 35% to 38 %) (fig 6B). These results suggest that Fas does not play a major role in H pylori induced apoptosis.
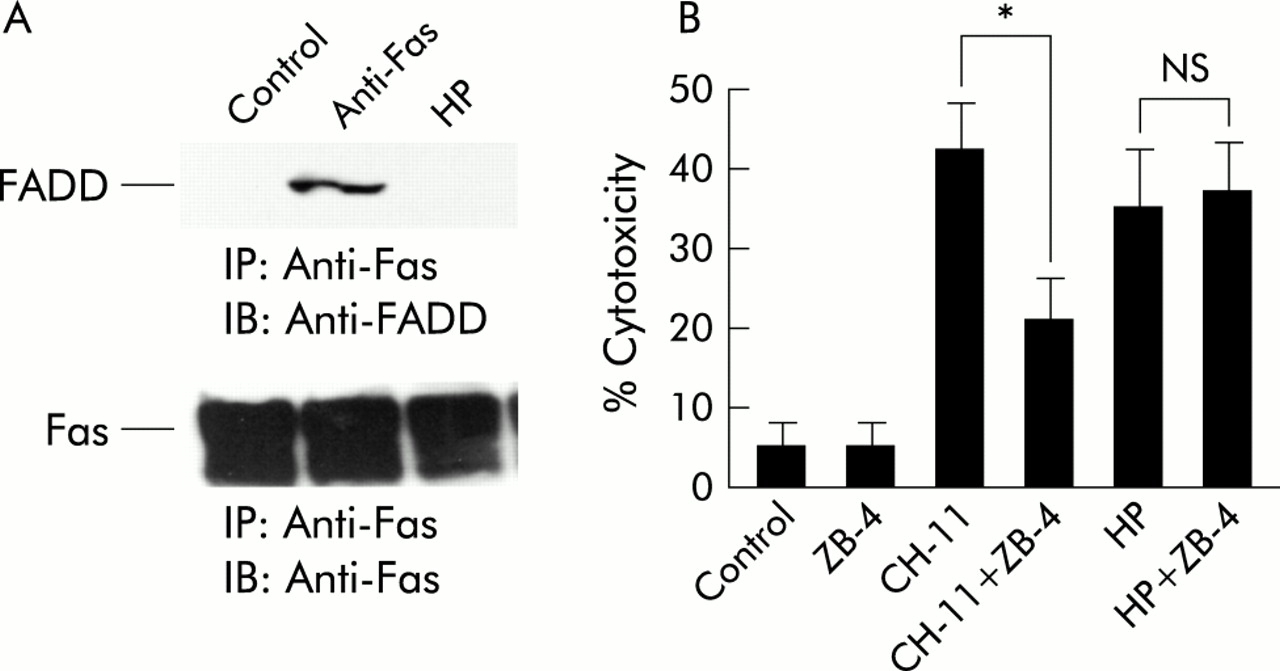
Fas (CD95) was not associated with Helicobacter pylori induced apoptosis. (A) MKN45 cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours. Cells were then incubated with H pylori (HP) or anti-Fas (CH-11) for 24 hours. Total cell lysates were extracted and immunoprecipitated with anti-Fas antibody and immunoblotted with anti-Fas associated death domain protein (FADD) and anti-Fas antibody. (B) MKN45 cells were treated with IFN-γ (10 ng/ml) for 24 hours, with or without neutralising anti-Fas antibody (ZB-4) for one hour, and incubated with H pylori or anti-Fas (CH-11). Cell viability was assessed by trypan blue dye exclusion assay at the indicated times by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of viable cells. Values are mean (SD) of three independent experiments. *Percentage cytotoxicity was significantly (p<0.05) different between the anti-Fas treatment group and the anti-Fas treatment with ZB-4 treatment group. NS, no significant difference was found.
H pylori induces cytochrome c release from the mitochondria
To examine whether or not H pylori induced apoptotic signalling involves a mitochondrial pathway, MKN45 cells were treated with H pylori, and mitochondria free cytosolic extracts were prepared and analysed by immunoblotting. Cytochrome c accumulated in cytosolic extracts after 12 hours of coculturing with H pylori. In examining the effects of the agonist anti-Fas antibody, as previously reported in the other cell lines, cytochrome c also accumulated after exposure to anti-Fas in MKN45 cells (fig 7A). Similar results were obtained using TMK-1 cells. To examine whether or not cytochrome c accumulation is caspase-8 dependent, cells were pretreated with caspase-8 inhibitor (Ac-IETD-CHO) for one hour before exposure to H pylori or anti-Fas. Although anti-Fas mediated cytochrome c release from the mitochondria was inhibited by the caspase-8 inhibitor, H pylori mediated release was not inhibited. We also used the pan-caspase inhibitor Z-VAD-FMK. Similar to the caspase-8 inhibitor, the pan-caspase inhibitor abolished anti-Fas mediated cytochrome c release from the mitochondria but not H pylori mediated release. This indicates that H pylori induced cytochrome c release from the mitochondria is not caspase-8 dependent (fig 7B).

Helicobacter pylori induced cytochrome c release from the mitochondria. (A) MKN45 cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours, and then with H pylori or anti-Fas (CH-11). At the indicated times, cytosolic fractions and total cell lysates were extracted, separated by electrophoresis, and immunoblotted with anti-cytochrome c and anti-actin, respectively. (B) MKN45 cells were treated with IFN-γ (10 ng/ml) for 24 hours, and 100 μM of the caspase-8 inhibitor Ac-IETD-CHO (IETD), the pan-caspase inhibitor Z-VAD-FMK (ZVAD), or medium alone pretreatment for one hour before exposure to H pylori (HP) or anti-Fas (CH-11). Cells were then incubated with H pylori or anti-Fas (CH-11) for 24 hours. Cytosolic fractions and total cell lysates were extracted, separated by electrophoresis, and immunoblotted with anti-cytochrome c and anti-actin.
Expression of apoptosis related proteins
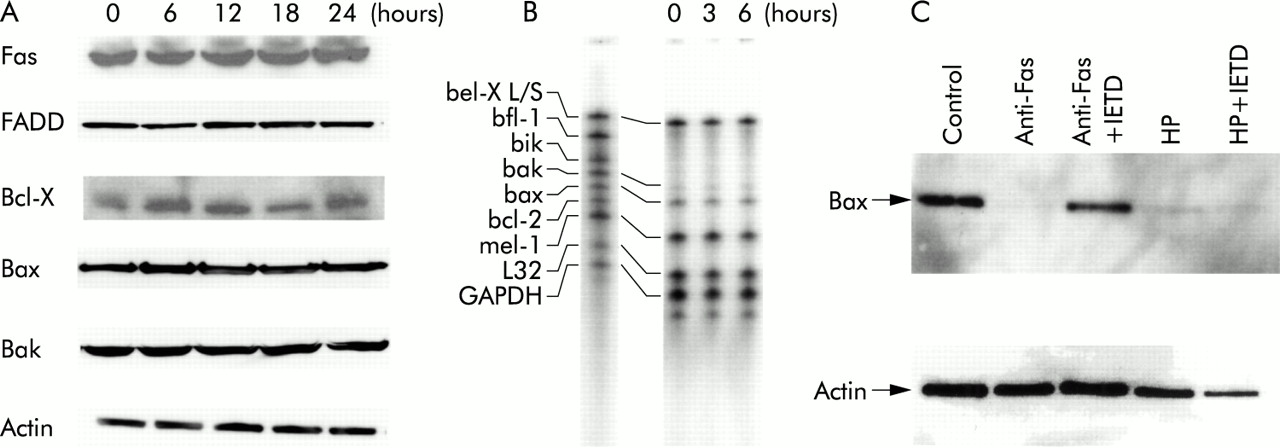
We determined whether H pylori mediated apoptosis correlated with expression of apoptosis related proteins. Coculture with H pylori cells did not change levels of Fas, FADD, Bax, Bak, or Bcl-X in total cell lysates extracted from MKN45 at the indicated times (fig 8A). Similar results were obtained using TMK-1 cells. We also evaluated mRNA expression of Bax, Bak, and Bcl-X using a ribonuclease protection assay and found no apparent changes. It is known that Bax may be translocated from the cytosol to the mitochondrial membrane without changing total cell amount. Thus we examined the immunoblot analysis of Bax using only the cytosolic fraction and found that cytosolic levels of Bax were decreased in H pylori mediated apoptosis as well as in Fas mediated apoptosis, indicating that Bax is translocated into mitochondria in H pylori mediated apoptosis (fig 8C). We also evaluated the effect of the caspase-8 inhibitor. The inhibitor abolished the decrease in cytosolic Bax induced by anti-Fas but did not affect the decrease induced by H pylori (fig 8C).
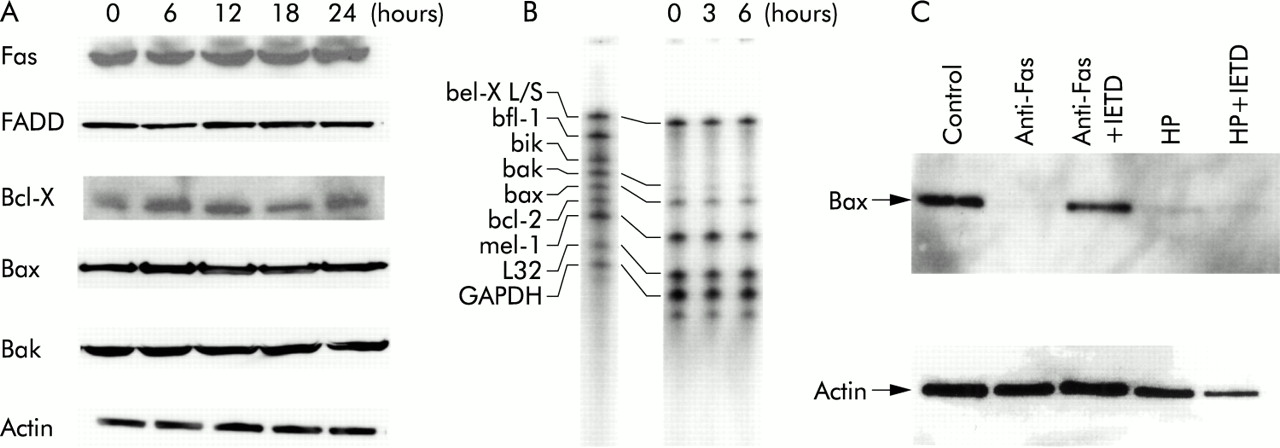
Expression of apoptosis related protein. (A) MKN45 cells were incubated with Helicobacter pylori and total cell lysates were extracted at the indicated times. The same amount of extracted protein (20 μg) was separated by electrophoresis and immunoblotted with anti-Fas, Fas associated death domain protein (FADD), Bcl-X, Bax, Bak, and actin. (B) MKN45 cell were incubated with H pylori and total RNA was extracted at the indicated times. The ribonuclease protection assay was performed according to the supplier's instructions. (C) MKN45 cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours, and 100 μM of the caspase-8 inhibitor Ac-IETD-CHO (IETD) or medium only pretreatment for one hour before exposure to H pylori (HP) or anti-Fas (CH-11). Cells were then incubated with H pylori or anti-Fas (CH-11) for 24 hours. Cytosolic fractions and total cell lysates were extracted, separated by electrophoresis, and immunoblotted with anti-Bax and anti-actin.
H pylori mediated NFκB activation suppresses apoptotic effects
We previously described cag PAI positive H pylori activation of NFκB in gastric cancer cells. There was a significant decrease in induction of NFκB transiently transfected with mutant IκBα (S32A/S36A) (super-repressor), indicating that phosphorylation of IκBα at Ser32 and Ser36 is critical for H pylori mediated activation of NFκB. To determine whether NFκB activation correlated with H pylori induced apoptosis, we generated stable transformants of MKN45 cells expressing mutant IκBα (MKN45 IκBα SR) (fig 9A). To examine the effect of stable expression of Iκβα super-repressor, we performed a electrophoretic mobility shift assay. H pylori and tumour necrosis factor α activated NFκB in MKN45-IκBα SR cells significantly less than in wild-type MKN45 cells. This result indicated that stable expression of Iκβα super-repressor construct is functioning as an inhibitor (fig 9B). Cell cytotoxicity caused by H pylori or the agonist anti-Fas was evaluated in the IκBα (S32A/S36A) transformants and compared with control cells. Both H pylori and the agonist anti-Fas significantly decreased the viability of the transformant cells at 12, 24, and 36 hours (fig 9C). To compare levels of DNA fragmentation, we used fragmentation ELISA at 12 hours after infection. DNA fragmentation was increased in MKN45 IκBα SR cells compared with MKN45 cells, indicating that specific apoptosis was increased in MKN45 IκBα SR cells (fig 9D). Caspase-8 and caspase-3, as revealed by immunoblot, were activated more rapidly (fig 9E). These results indicate that NFκB activation by H pylori demonstrated antiapoptotic effects. To evaluate expression of antiapoptosis related genes, we performed a ribonuclease protection assay. C-IAP1 and IAP2 were upregulated by H pylori infection in MKN45 cells. cagE mutant H pylori demonstrated a smaller potential to upregulate these genes (fig 9F). These results indicate that H pylori may exert antiapoptotic effects by upregulating c-IAP genes through NFκB activation.
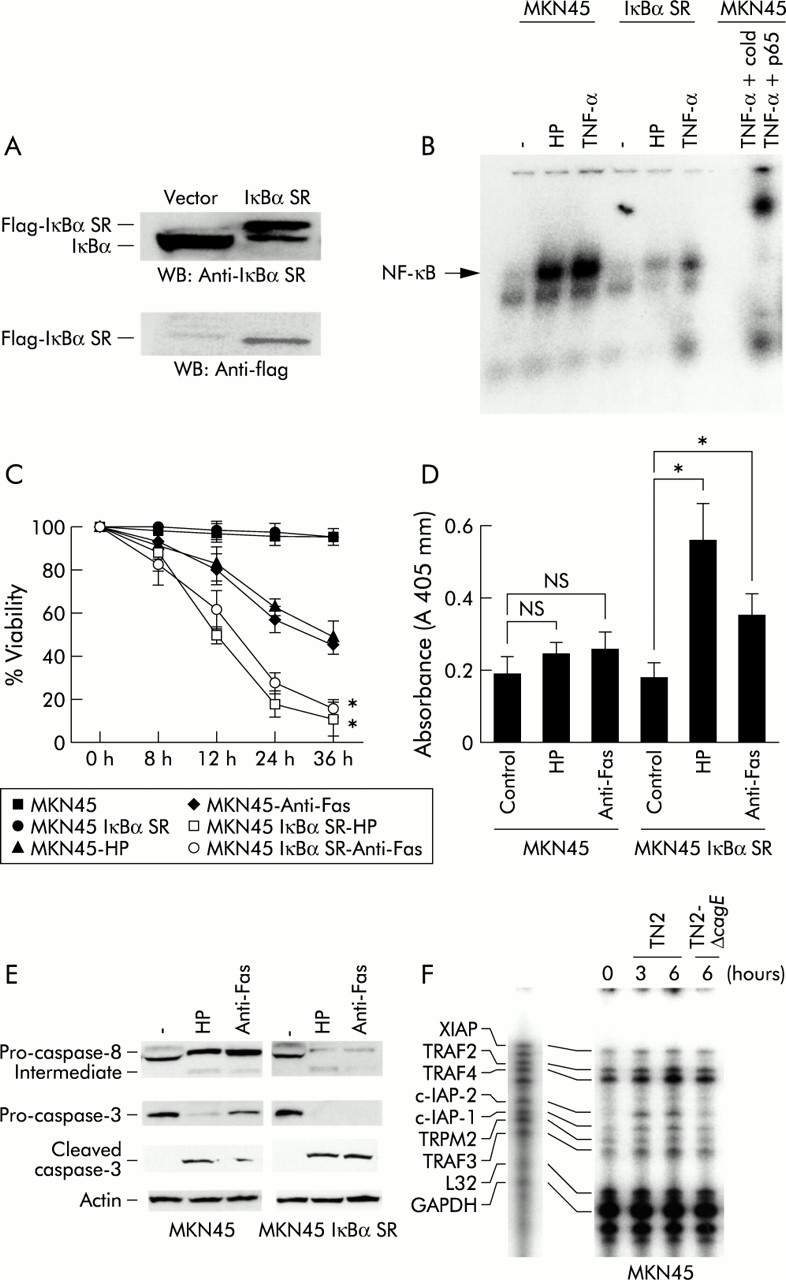
(A) Detection of stable MKN45 transfectants expressing Iκβα (SS32/36AA). The clone was analysed using a monoclonal antibody to FLAG and polyclonal antibody to Iκβα. (B) Specific protein binding activities of nuclear factor kappa B (NFκB) sequences (electrophoretic mobility shift assay). The nuclear extracts were prepared from MKN45 and MKN45 IκBα SR cells. Cells were treated or not treated with Helicobacter pylori (HP) or 10 ng/ml tumour necrosis factor α (TNF-α) for 90 minutes. Nuclear extracts was incubated with 32P labelled oligonucleotide for 30 minutes. Migration of the DNA-protein complex containing NFκB is indicated. This complex was found to be specific, as judged using supershifting antibody against p65 and cold NFκB probes. (C) MKN45 and MKN45 Iκβα SR cells were treated with interferon γ (IFN-γ 10 ng/ml) for 24 hours. Cells were incubated with H pylori (HP) or anti-Fas (CH-11) for 24 hours. Cell viability was assessed by trypan blue dye exclusion assay by counting 300 cells. Viable cells were quantitated under bright field microscopy. Results are expressed as percentages of dead cells. Values are mean (SD) of three independent experiments. *Percentage cytotoxicity was significantly (p<0.05) different between the MKN45 treated cells with anti-Fas or cocultured with H pylori and MKN45 IκBα SR treated cells with anti-Fas or cocultured with H pylori. (D) DNA fragmentation was quantified using a commercially available ELISA (Boehringer Mannheim Biochemicals, Mannheim, Germany): 5×104 cells were incubated in triplicate with H pylori (HP), anti-Fas (CH-11), or medium alone for 12 hours and lysed, and the supernatants were used for ELISA. Absorbance was measured at 405 nm. *Absorbance was significantly (p<0.05) different between the control and treated with anti-Fas or cocultured with H pylori in the MKN45 IκBα SR groups; NS, no significant difference was found. (E) MKN45 and MKN-45 IκβαSR cells were treated with IFN-γ (10 ng/ml) for 24 hours. Cells were incubated with H pylori (HP) or anti-Fas (CH-11) for 12 hours. Immunoblot analysis was performed using anti-caspase-8, caspase-3, cleaved caspase-3, and anti-actin antibody. (F) MKN45 and THP-1 cells were incubated with H pylori and total RNA was extracted at the indicated times. The ribonuclease protection assay was performed according to the supplier's instructions.
DISCUSSION
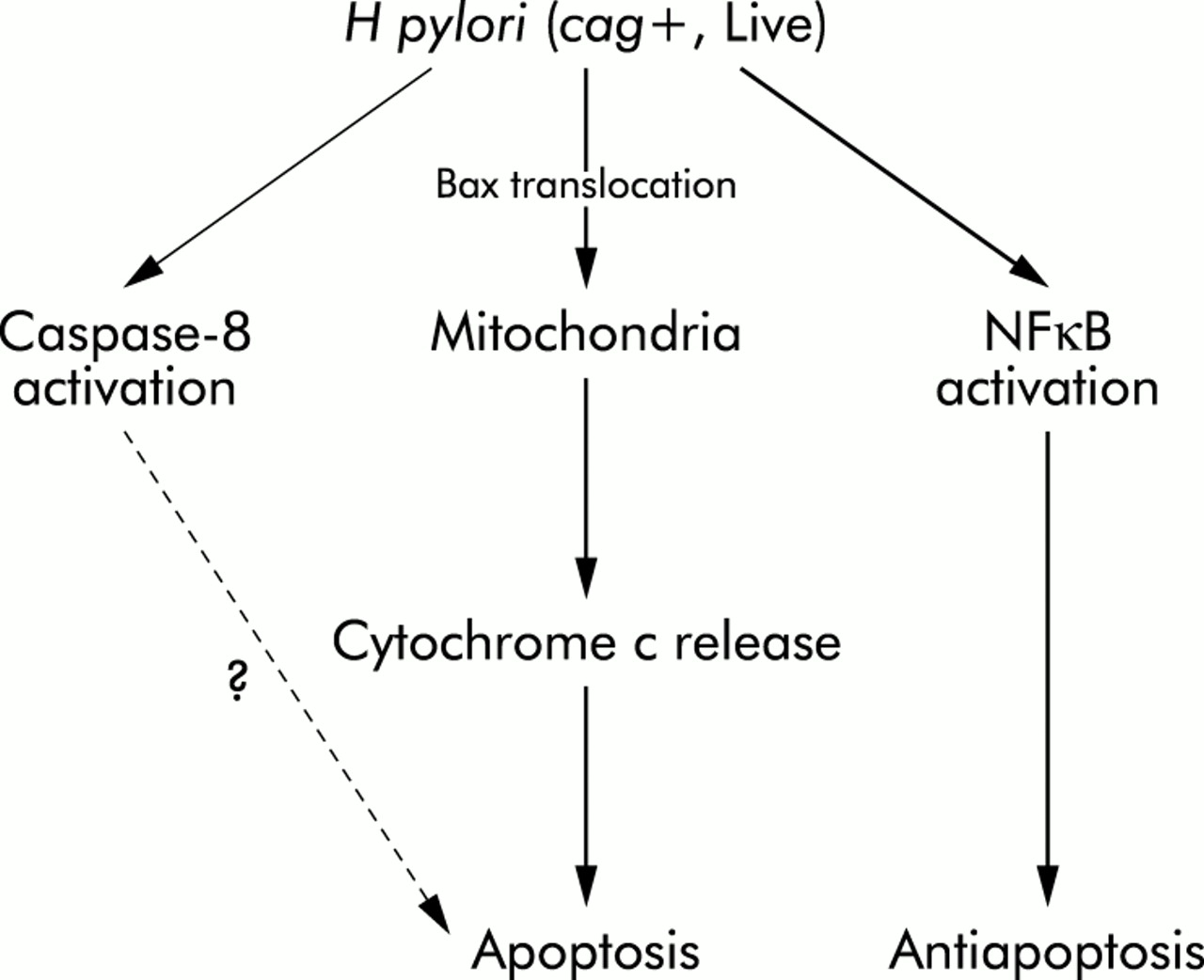
In this study we have shown that cytochrome c release from the mitochondria is a major pathway in H pylori mediated apoptosis. Although caspase-8 was indeed activated, the caspase-8 inhibitors did not affect H pylori mediated apoptosis, indicating that the major pathway is independent of caspase-8 activation (fig 10).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Schematic representation of the signalling pathways leading to apoptosis and antiapoptosis in response to Helicobacter pylori in gastric epithelial cells.
Activation of caspase-8 usually occurs as a consequence of surface receptor binding to its ligand. Reportedly, the Fas-Fas ligand system is involved in H pylori mediated apoptosis in vivo and in vitro10,16,17 but in this study neutralising anti-Fas antibody did not affect H pylori mediated apoptosis in cultured cancer cells. Moreover, we found no evidence of recruitment of FADD to Fas, a downstream event of Fas-Fas ligand association, in H pylori mediated apoptosis. Thus involvement of the Fas-Fas ligand system in H pylori mediated apoptosis was not substantiated, and the mechanism of caspase-8 activation has yet to be clarified. In addition, the caspase-8 inhibitor Ac-IETD-CHO did not inhibit H pylori mediated apoptosis, suggesting that caspase-8 dependent signal transduction does not play a major role in H pylori mediated apoptosis, at least in vitro. Although it is reported that the Fas-Fas ligand system is important in vivo, activation of this pathway may be induced indirectly and secondary. We suggest that H pylori is capable of inducing apoptosis mainly through the mitochondrial pathway in vitro. The importance of this pathway in vivo remains unknown.
Various proapoptotic signals, such as those induced by radiation, anticancer drugs, and stress, converge at cytochrome c release from the mitochondria. Although the current data suggest that cytochrome c release also plays a pivotal role in H pylori mediated apoptosis, the exact mechanism has yet to be investigated. The status of Bcl-2 family proteins determines whether a cell will live or die through regulation of cytochrome c release from the mitochondria.13–15 Increased Bak expression, a proapoptotic member of the Bcl-2 family, in H pylori infection was recently reported.18 However, we did not find changes in expression of Bax, Bak, or Bcl-X in either mRNA or protein levels when cocultured with H pylori. These findings do not exclude the possibility that some Bcl-2 family proteins are involved in H pylori mediated apoptosis. For example, Bax may translocate from the cytosol to the mitochondria for integration into the membrane following a proapoptotic stimulus. This action then results in cytochrome c release while the total amount of protein remains constant.34 Thus we checked by immunoblot analysis the level of Bax in the cytosolic fraction and found that cytosolic Bax was decreased in H pylori mediated apoptosis as well as in Fas mediated apoptosis, suggesting that Bax was translocated into mitochondria in H pylori mediated apoptosis.
p53 is an important molecule that affects apoptosis.35 Bax gene expression caused by p53 may be an important part of p53 mediated apoptosis.36 Reportedly, H pylori infection in patients resulted in nuclear staining for p53 in the glandular cells of the mucosa, and bacterial eradication caused a decrease in p53 accumulation in epithelial cells.37–39 However, we did not find upregulation of Bax or MDM2, which are regulated by p53 in cells with normal p53, including MKN45 cells, suggesting that H pylori cannot activate p53 directly in vitro (unpublished observation).
We have previously shown that H pylori induces NFκB activation. In this study, we confirmed that H pylori mediated NFκB activation exerts antiapoptotic effects in MKN45 cells where upregulation of c-IAP1 and 2 may be involved. Thus H pylori has both apoptotic and antiapoptotic effects. Similarly, the cytokine tumour necrosis factor α can produce bidirectional effects on apoptosis. The signal triggered by tumour necrosis factor α binding to its receptor bifurcates at TRADD: one signal induces NFκβ activation via RIP promoting cell survival and the other induces apoptosis via FADD.40,41
The adapter molecule myeloid differentiation factor 88 (MyD88) mediates both apoptosis and NFκB activation through Toll-like receptors.42,43 Inhibition of the NFκB pathway downstream of MyD88 potentiates apoptosis, indicating that these two pathways bifurcate at the level of MyD88. Moreover, MyD88 binds FADD and is sufficient to induce apoptosis. Recently, we revealed that NFκB activation caused by H pylori was associated with TRAF6, a molecule downstream of MyD88.32 In addition, as H pylori is a gram negative bacterium, the relation to Toll-like receptors should be investigated.
Knockout of cagE, one of the cag PAI genes, decreased both the apoptotic effects and potential to activate NFκB. Thus it may be speculated that both apoptosis and NFκB activation are triggered by the same signal that is dependent on cag PAI. Several cag PAI proteins, including CagE, are thought to constitute a type IV secretion system. CagE itself is a homologue of a transporter component in Agrobacterium tumefaciens and Bordetella pertussis that engage in the transcellular transport of toxins or T-DNA.44,45 Recent reports suggest that the system transports CagA protein, another product of the cag PAI, into the cytoplasm of host cells, where CagA undergoes tyrosine phosphorylation.46,47 As the system may also transport other molecules, it is possible that a certain protein produced by H pylori is transported into host cells where it triggers both apoptotic and NFκB activating signalling pathways. Recently, we have revealed that knockout of the cagE gene deprived wild-type H pylori on the pathogenicity for gastric ulcer, gastritis, and intestinal metaplasia in an in vivo model.48 This observation may indicate that the pro- and antiapoptotic effect induced by wild-type H pylori is an important factor in the pathogenesis of H pylori mediated gastric diseases.
In conclusion, we demonstrated that H pylori directly induced apoptosis mainly through cytochrome c release from the mitochondria. H pylori also exerted antiapoptotic effects through NFκB activation. Both apoptotic and antiapoptotic effects were dependent on cag PAI.
Acknowledgments
We are grateful to Dr H Suzuki for providing us with the super-repressor mutant of IκBα (SS32/36AA). We would also like to thank Ms Mitsuko Tsubouchi for her excellent technical assistance. This study was supported in part by a grant-in-aid for Scientific Research from the Ministry of Education, Science, Sports, and Culture 11557040.