Article Text

Abstract
The Epstein-Barr virus (EBV) infects humans and the genome of this infectious agent has been detected in several tumour types, ranging from lymphomas to carcinomas. The analysis of the functions of the numerous viral proteins encoded by EBV has been impeded by the large size of the viral genome, which renders the construction of viral mutants difficult. To overcome these limitations, several genetic systems have been developed that allow the modification of the viral genome. Two different approaches, depending on the host cell type in which the viral mutants are generated, have been used in the past. Traditionally, mutants were constructed in EBV infected eukaryotic cells, but more recently, approaches that make use of a recombinant EBV cloned in Escherichia coli have been proposed. The phenotype associated with the inactivation or modification of nearly 20 of the 100 EBV viral genes has been reported in the literature. In most of the reported cases, the EBV latent genes that mediate the ability of EBV to immortalise infected cells were the targets of the genetic analysis, but some virus mutants in which genes involved in DNA lytic replication or infection were disrupted have also been reported. The ability to modify the viral genome also opens the way to the construction of viral strains with medical relevance. A cell line infected by a virus that lacks the EBV packaging sequences can be used as a helper cell line for the encapsidation of EBV based viral vectors. This cell line will allow the evaluation of EBV as a gene transfer system with applications in gene therapy. Finally, genetically modified non-pathogenic strains will provide a basis for the design of an attenuated EBV live vaccine.
- Epstein-Barr virus
- gene therapy
Statistics from Altmetric.com
The Epstein-Barr virus (EBV) together with Kaposi's sarcoma herpesvirus (KSHV, also termed human herpesvirus 8; HHV-8), belongs to the subgroup of γ-herpesviruses. These viruses have been detected in different human malignant tumours and recently EBV has been incorporated into the group of oncogenic viruses.1 Most of the human population is infected with and carries the genome of EBV for a lifetime without clinical symptoms, implying that this virus exerts its oncogenic potential only under particular conditions. The same observation holds true for HHV-8, the most recently described member of the γ-herpesviruses, which is endemic in some regions of the world, such as southern Italy and Africa, and rarely leads to Kaposi's sarcoma in infected individuals. One of the most challenging problems is the identification of the circumstances and conditions that lead to the development of tumours in the context of EBV or HHV-8 infection. Patients with immunodeficiency, either congenital or acquired (for example after immunosuppressive treatment for organ transplantation or human immunodeficiency virus (HIV) infection) have a much higher risk of developing EBV or HHV-8 associated tumours than the general population, pointing to the crucial role of the immune system in controlling the proliferation of EBV or HHV-8 infected cells.
The EBV genome has been detected in a variety of tumours, including Hodgkin's disease, some non-Hodgkin's lymphomas, Burkitt's lymphoma, nasopharyngeal carcinoma, certain T cell lymphomas, and several other malignancies. For most of these tumours no predisposing conditions are known.1 Similarly, the role of HHV-8 in the development of unusual body cavity lymphomas or Kaposi's sarcoma is equally unclear. Whether these viruses are true aetiological factors in human malignancies or whether they contribute to one of the numerous steps that lead to transformation is an important question that is still unanswered.
A unique characteristic of EBV is its ability to infect resting human B cells in vitro. After infection, the virus establishes a latent infection, during which the expression of the so called latent genes sustains an indefinite proliferation of the infected B cells. In general, no virus is produced during latent infection, but in a certain proportion of EBV infected cells a reactivation of the viral lytic programme can be observed, with the release of mature infectious virions.2 The different viral programmes, which include the infection of target cells, immortalisation, the reactivation of the lytic phase of EBV's life cycle including DNA replication, the expression of viral structural proteins, and virion maturation, are controlled by nearly 100 viral genes, making EBV one of the more complex viruses. This complexity is reflected in the large size of the viral genome, encompassing about 170 kb DNA. As expected, many gene products are used at different stages of viral infection and constitute a discretely regulated and hierarchical cascade, which renders the dissection of their functions difficult. As a consequence, even if the study of an isolated gene can give interesting clues about its functions in vivo, a deeper understanding requires its analysis in the context of the whole viral genome. Ideally, the mutation of a single gene within the whole viral genome is most informative because it can be compared directly with the corresponding wild-type virus. Despite the fact that the manipulation of large DNA molecules in eukaryotic cells is demanding, several systems for the genetic analysis of large viral genomes have been described.
Herpesviruses also possess several features that render them attractive for the design of viral vectors as tools for gene transfer in in vitro or in vivo settings. Several characteristics of herpesviruses, including their persistence in the host, their extrachromosomal replication in infected cells, and their large genomes, offer a variety of advantages when compared with other systems, such as retroviruses or adenoviruses. Hence, EBV derived vectors could allow the incorporation of up to 140 kb of foreign DNA, leaving enough space for the incorporation of large regulatory sequences often present within cellular genetic loci.
In this article, the different genetic systems that have been developed during the past years will be reviewed, as well as some of the applications of this technology. This includes information about the physiology of the viral infection as well as the use of mutated viral genomes for gene transfer experiments.
Experimental systems for the genetic analysis of EBV
CONSTRUCTION OF VIRAL MUTANTS IN EUKARYOTIC CELLS
Because of the large size of their genomes, herpesviruses have not been amenable to conventional recombinant DNA technology. Therefore, for many years homologous recombination was the method of choice to alter herpesvirus genomes genetically. Historically, various gene transfer methods were used extensively to introduce mutations into the viral genome. In herpesvirus infected cells, transient introduction of a DNA fragment that carries the mutated version of a gene of interest can lead to recombination events that target the wild-type copy of the gene. Recombination is promoted by flanking regions that enable the precise exchange of the wild-type versus the mutant allele. The addition of a selection marker—for example, a resistance gene in conjunction with the mutant allele, allows the selection of herpesvirus infected cells that propagate the genetically altered herpesvirus. This basic method, which was initially established in prokaryotic systems, has been extended successfully to members of the α-herpesvirus subfamily, herpes simplex virus in particular.3,4 The high efficiency of this method for α-herpesviruses relies on the massive viral DNA replication that takes place in the infected cells. In addition, viral DNA replication stimulates DNA recombination in general in the infected cells, facilitating the generation of viral mutants. Because mutant viruses are frequently admixed with wild-type ones, it is necessary to purify the recombinant virus stocks. To this aim, suitable eukaryotic cells are infected with diluted viral stocks containing wild-type viruses admixed with their mutant counterparts, such that only a few cells become infected by a single virion. The isolation of plaques that result from viral infection allows the enrichment of a single virus. Additional rounds of purification enable the isolation of mutated viral strains devoid of any contaminating wild-type herpesvirus. This procedure can at times be tedious, particularly when the mutation confers a replication disadvantage compared with wild-type virus. Obviously, this approach will be unsuccessful in the case of mutations that target genes indispensable for virus propagation in vitro.
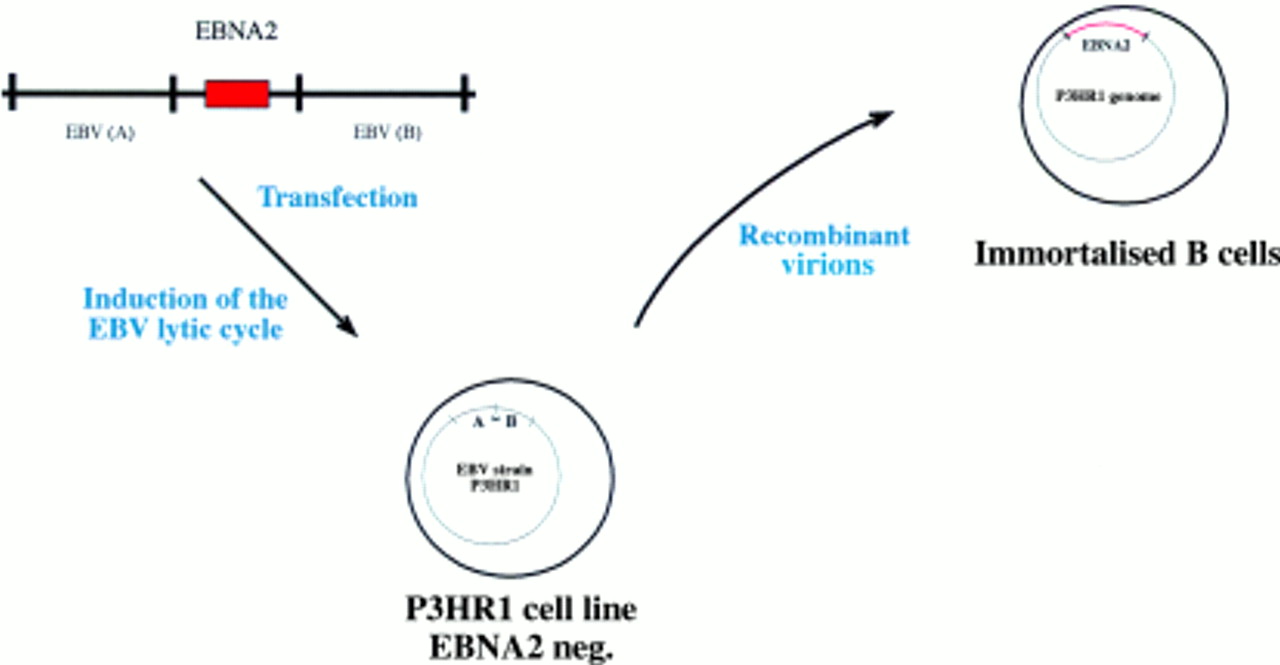
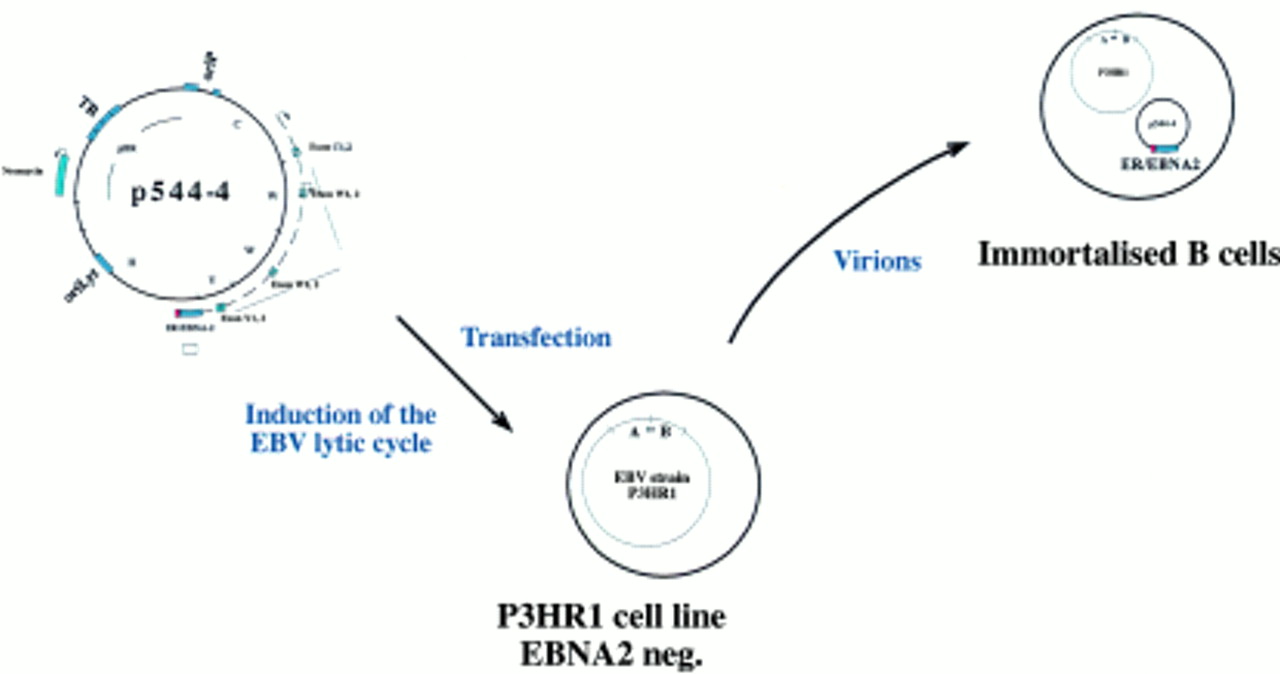
This procedure has also been applied successfully to EBV, although the recombination efficiency in the case of EBV is several orders of magnitude lower than with the α-herpesviruses. In addition, separation of the recombinant from wild-type viruses is much more difficult because no cell can be infected with EBV to give rise to progeny virus. As alternative approaches, two methods have been developed for the isolation of recombinant EBVs. Initially, the immortalising characteristics of EBV have been used to select and purify recombinant versus wild-type EBV. In the first two published reports of the successful construction of a recombinant EBV, the EBV encoded nuclear protein 2 (EBNA2) gene was reintroduced into the genome of the P3HR1 viral strain, a laboratory strain in which the EBNA2 gene is deleted.5,6 P3HR1 is incapable of immortalising B cells because EBNA2 is absolutely required for this process.7,8 Recombinant P3HR1 genomes into which EBNA2 had been introduced regained their immortalising potential (fig 1). Infection of primary B cells led to permanently proliferating cell lines that harbour the recombinant EBV genome.5,6 A variation on this theme consisted of a technique in which the missing EBNA2 gene is provided in a plasmid that can be packaged into a viral coat.6 As an extension to this approach, a conditional EBNA2 allele was established with which the proliferation of B cells can be initiated or stopped at will. To this aim, target cells are coinfected with both the P3HRI virus and a plasmid that carries a wild-type EBNA2 gene,6 or a fusion between the EBNA2 gene and the oestrogen receptor.9 Because the plasmid carries both EBV replication origins and the EBV packaging sequences, it is packaged into a viral particle and maintained as an episomal copy in the infected cell (fig 2). The fusion between EBNA2 and the oestrogen receptor allows a precise evaluation of the viral and cellular targets of the EBNA2 gene. The EBNA2 transactivator is absolutely required for B cell immortalisation and is active only in the presence of oestrogen. Withdrawal of oestrogen leads to a reversible arrest in cell growth.9 After the reintroduction of oestrogen, the direct target genes of EBNA2 can be identified.
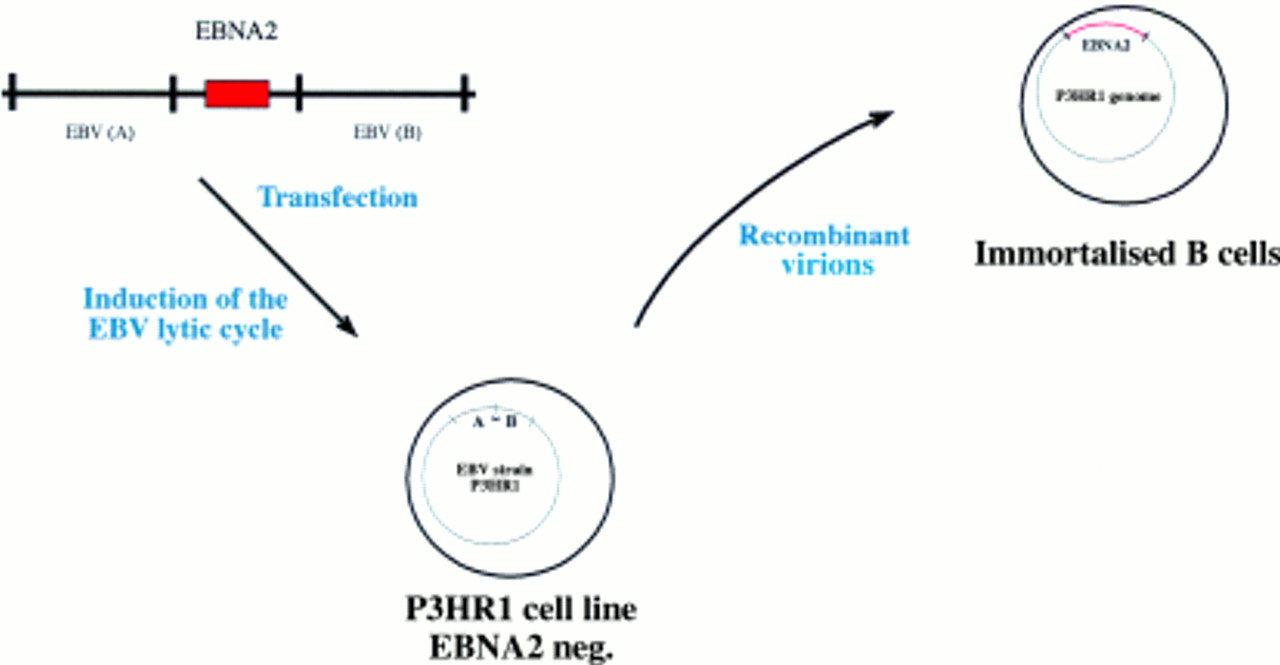
The Epstein-Barr virus (EBV) genetic system in eukaryotic cells (I). The P3HR1 EBV strain harbours a deletion in the EBV encoded nuclear protein 2 (EBNA2) gene locus that renders it non-immortalising. A plasmid that carries the EBNA2 gene, cloned between EBV homologous sequences that flank it in the wild-type EBV genome, is transfected into the P3HR1 cell line. This transfection leads to the recombination of the plasmid with the resident P3HR1 genome. The recombinant viruses can be positively selected owing to their capacity to immortalise primary B cells.

The oestrogen responsive Epstein-Barr virus (EBV) encoded nuclear protein 2 (EBNA2) gene provides a conditional system for the study of EBV mediated B cell immortalisation. This system is based on coinfection of primary B cells by the P3HR1 virus and a plasmid that carries EBNA2. As an alternative, the EBNA2 gene was fused to the oestrogen receptor (ER) to generate a conditional EBNA2 allele with which the latent EBV programme can be activated or inhibited by the addition or withdrawal of oestrogen, respectively.
Recombination of the P3HR1 genome with DNA fragments that contain EBNA2 in conjunction with adjacent viral genes of interest allowed an extension of the recombination method to other genes (fig 3). Transfection of partially overlapping cosmids that encompass the whole viral DNA will recombine to produce a complete herpesvirus genome. In principle, this approach allows one to target any viral gene as long as the constructed mutant can be propagated.10 In these methods, however, negative selection against wild-type viruses is not used and viral supernatants contain both wild-type and recombinant viruses. Therefore, EBV target cells need to be infected under experimental conditions that lead to the infection of target cells with a single virus. Limiting dilution allows the isolation of EBV infected and immortalised cell clones that contain the recombinant virus exclusively. Eventually, immortalised B cell clones that are latently infected by the recombinant viral episome can be induced to produce viral stocks that contain virions with the mutated viral genome only. The success of this purification step depends on the ability of the recombinant mutants to replicate, become encapsidated, and give rise to immortalised B cell clones. This purification step therefore excludes virions that carry mutated replication origins or other cis-acting sequences required for encapsidation of the viral DNA or for virus maturation and assembly. Moreover, immortalised B cell lines are frequently difficult to induce so that supernatants with high virus titres are rarely obtained. Finally, because the amount of virus produced in these cells is not high enough to allow an easy evaluation of the viral DNA, it remains difficult to exclude minimal modifications of the viral DNA that could have occurred during recombination.

The Epstein-Barr virus (EBV) genetic system in eukaryotic cells (II). The selection system that makes use of the EBV encoded nuclear protein 2 (EBNA2) negative P3HR1 viral strain allows the incorporation of mutations into adjacent viral genes. A plasmid that carries the EBNA2 gene as well as a mutated allele of a viral gene is introduced into the cell line that carries P3HR1. Only viruses that have successfully recombined with this plasmid recover the capacity to immortalise B cells. This recombination event also promotes the incorporation of the mutant allele into the viral genome.
An alternative approach has relied on introducing a selection marker (for example, the neomycin or the hygromycin resistance gene) into the viral genome.11,12 The drug resistance cassette is flanked by specific EBV sequences that allow the precise incorporation of the selection marker into the viral genome. This construct can be designed in such a way that its incorporation leads to the disruption of a viral gene to yield a knockout viral mutant. After the introduction of the selection marker cassette into an EBV infected cell line and activation of the lytic cycle, supernatants contain wild-type as well as mutant viruses. EBV negative Burkitt's lymphoma cell lines (or Burkitt's lymphoma cell lines that have lost the virus during propagation in vitro) are frequently used as target cells for the viral infection with recombinant virus stocks.13–15 After drug selection, cell clones that contain the mutant virus only can be isolated and used for the production of pure mutant virus stocks. As is the case with immortalised B cell lines, the inducibility of the selected clones is generally low. For this reason, the Akata cell line derived from a case of Burkitt's lymphoma has been used frequently for viral production because this cell line yields unusually high viral titres after induction.16
CONSTRUCTION OF VIRAL MUTANTS IN ESCHERICHIA COLI
In the strategies described above, all steps required for the construction of viral mutants, including the recombination between the viral genome and the plasmid that carries the DNA fragments to be incorporated, take place in eukaryotic cells. Ideally, the construction of viral mutants should be performed in bacterial cells because prokaryotic organisms are much easier to manipulate genetically than their eukaryotic counterparts. A prerequisite for such a strategy is the introduction of a prokaryotic replicon into the viral genomic DNA that allows propagation in a prokaryotic host. Another potential advantage of this technology relies on the assumption that any genetic alteration can then be introduced into the herpesvirus genome because it acts as a non-functional piece of DNA in a prokaryotic host. Introducing loss of function modifications of elements required for lytic replication or virion maturation or assembly is possible in a prokaryotic but not in a eukaryotic system. This is because the complete lytic phase of the EBV life cycle is a prerequisite for the separation of recombinant from wild-type virions in the latter system. As a consequence, we have established a prokaryotic system for the genetic analysis of EBV functions.
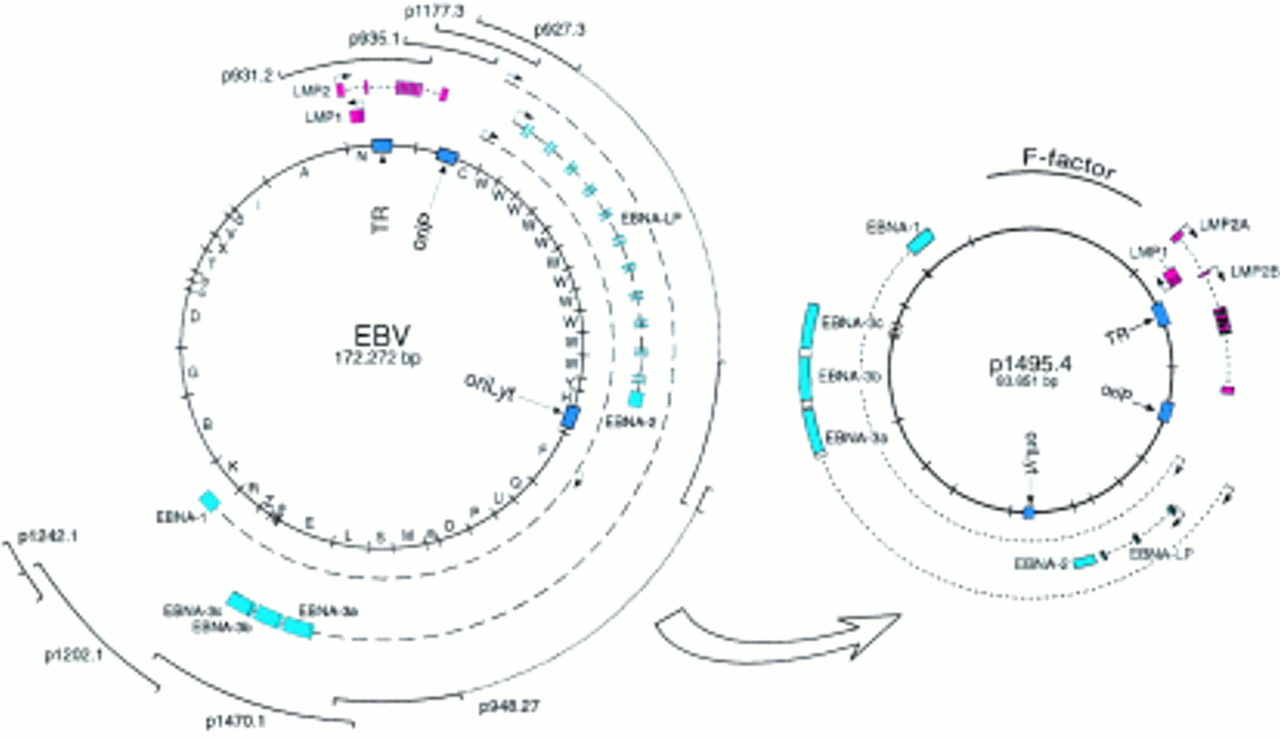
Our initial method to modify the functions of EBV in E coli relied on so called mini-EBVs. These artificial subgenomic EBV plasmids were constructed by the sequential addition of large pieces of viral DNA into a derivate of the F-plasmid (fig 4).17 The F-plasmid, a natural host of Gram negative bacteria, is a single copy plasmid in E coli characterised by superior stability, despite its large size of up to 300 kb.18 Less than 10 kb of a mini-F-plasmid is required for its faithful replication in E coli, leaving enough space for the incorporation of the complete genomes of any of the known herpesviruses. The construction of these mini-EBVs is based on multiple rounds of the chromosome building technique that uses temperature sensitive shuttle plasmids.18 In this method, the DNA fragment to be added to the F-plasmid is cloned into an intermediate shuttle plasmid that will transfer the new DNA fragment to the pre-existing viral part of the F-plasmid. As a first step, both the shuttle and the F-plasmid are recombined to form a cointegrate that will be resolved in a second step to eliminate the entire shuttle plasmid. This sequential process can be performed an almost unlimited number of times, although the growing size of the F-plasmid after each additional step makes the F-plasmid progressively more difficult to manipulate. The mini-EBV plasmid has been engineered up to half the size of the EBV genome and has been used to analyse EBV latent genes involved in B cell immortalisation. Because most EBV genes required for virus formation are absent from the mini-EBV genome, its encapsidation into a viral coat has to rely on a cell line containing an EBV helper virus, which will provide all viral factors required for the completion of the lytic cycle and virus maturation. Traditionally, the HH514 cell line, which is a subclone of the P3HR1 laboratory cell line, has been used for this purpose. This viral strain, also called P3HR1 (or HH514), carries a deletion of the EBNA2 gene that renders it incapable of immortalising primary human B cells. Mini-EBV plasmids can be packaged into an EBV coat after the transfection of the mini-EBV plasmid construct into HH514 cells and concomitant activation of the lytic cycle.17 However, because the EBV helper virus can also be encapsidated, supernatants contain infectious particles that carry the mini-EBV in conjunction with the helper virus. Human primary B cells are then infected with the mixed virus stocks. In a secondary step, immortalised cells infected with the mini-EBV only must be distinguished from cells infected with both the mini-EBV and the helper virus. Alternatively, the mini-EBV plasmid DNA purified from E coli can be transfected directly into primary B cells to give rise to immortalised B cell clones at a very low frequency, as a result of the low transfection rate of primary B cells.19 Because the mini-EBV contains only parts of the viral genome, not all viral functions can be analysed with a single construct.
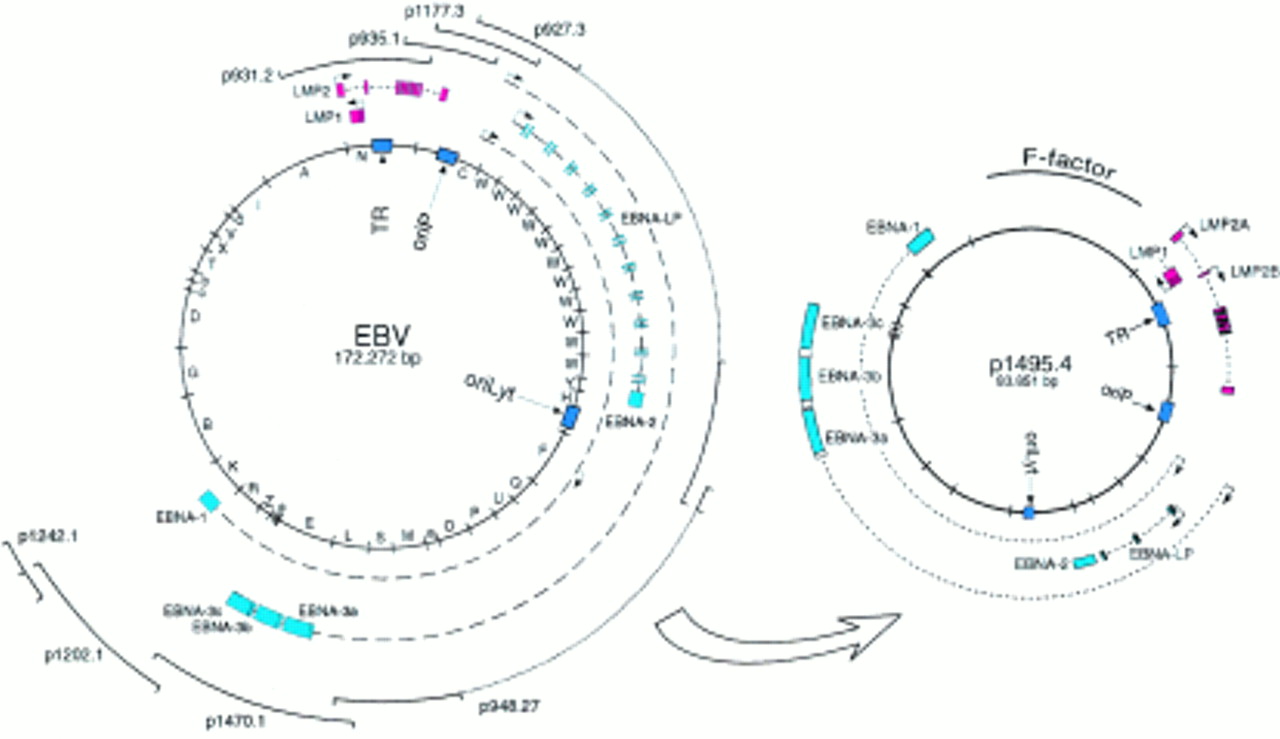
Epstein-Barr virus (EBV) genetic analysis in Escherichia coli: construction of the mini-EBV plasmid. Fragments from the EBV genome were sequentially added to the F-plasmid backbone using the chromosomal building technique. This mini-EBV carries all the latent genes and can immortalise primary B cells after encapsidation in a helper cell line or direct transfection of the plasmid. EBNA, EBV encoded nuclear protein; LMP, latent membrane protein.
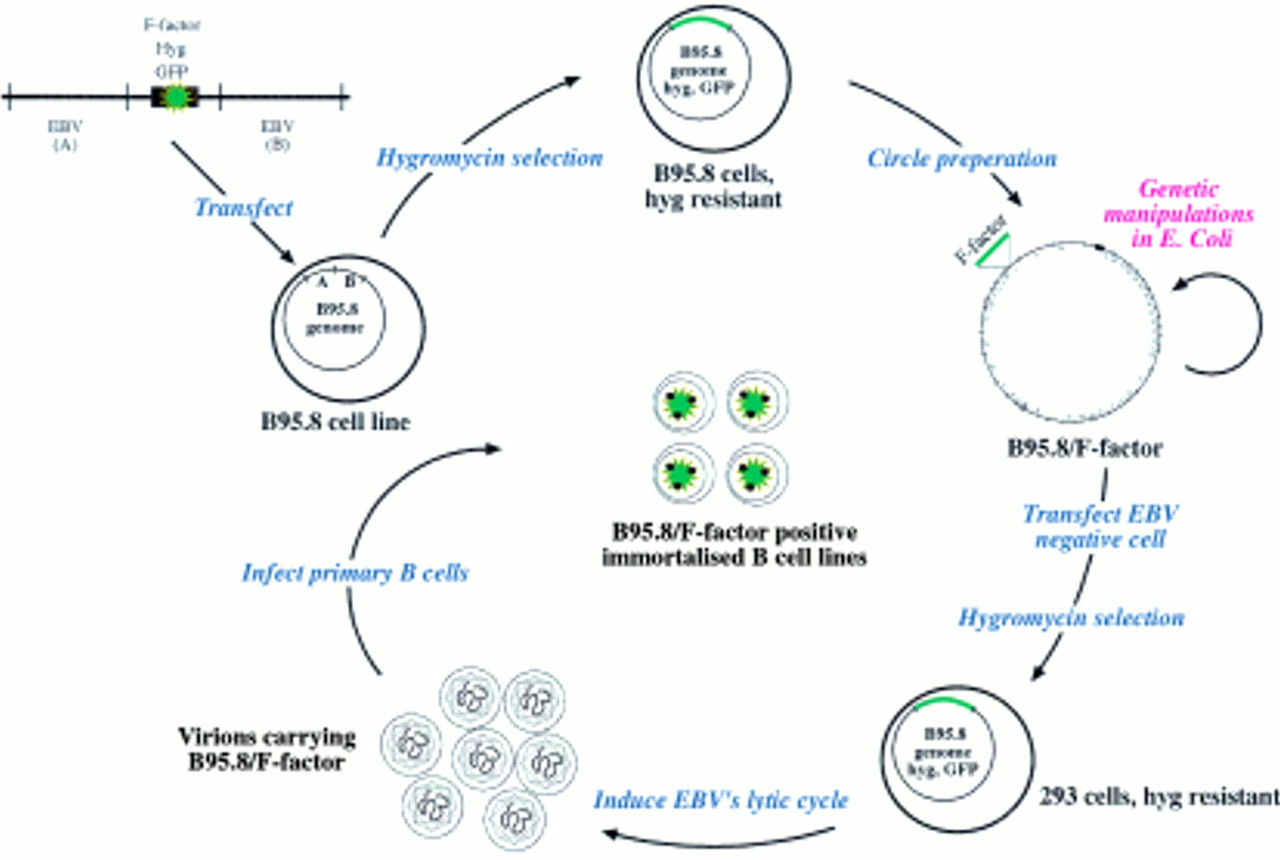
To extend the range of EBV genes amenable to genetic analysis, the initial technology was altered such that the F-plasmid replicon was introduced into the complete EBV genome to clone it as a whole in E coli.20 To this aim, an F-plasmid flanked with EBV sequences situated left and right from the deletion present in the EBV laboratory strain B95.8 was transfected into the cell line also termed B95.8, which is persistently infected with this particular EBV isolate (fig 5). Because the hygromycin resistance gene and the gene encoding the green fluorescent protein (GFP) were added to the F-plasmid based construct, the selection and identification of the viral molecules that successfully underwent homologous recombination with the transfected plasmid was possible. After bulk extraction of circular molecules present in the hygromycin resistant cell clones, the purified DNA from the recombinant viral DNA was used for subsequent transformation of the E coli strain DH10B. The recombinant DNA, which consisted of the B95.8 viral DNA and the F-plasmid, was found to be stable in E coli and could be propagated in the prokaryotic host. The cloned viral DNA can then be modified in E coli using either the technique of chromosomal building or, more directly, targeted allelic disruption.18,21,22 After modification in E coli, the viral DNA can be isolated and used to transfect EBV negative cells that support the lytic cycle. In this regard, 293 cells proved to be adequate, the virus titres obtained with this cell line being close to those obtained with the B95.8 cell line, which is frequently used to produce virions for the immortalisation of human B cells.20 The presence of the GFP gene on the EBV genome identifies the infected cells directly and also gives an estimate of the viral titre of a given viral stock. Raji is an EBV positive Burkitt's lymphoma cell line that can be (super-)infected with our recombinant virus. Thus, simply by counting the number of green cells obtained after infection of a defined number of Raji cells with the recombinant GFP encoding virus stock allows an estimate of its concentration. This system is particularly useful for the quantitative assessment of the consequences of different mutations in a single gene. Mutations are established independently and the virus concentration in different EBV mutant virus stocks must be adjusted to achieve comparable conditions. Using the Raji cell system, two viral populations, such as wild-type or mutated EBV, can be compared directly. Finally, because the viral DNA can be purified from E coli, transfection of the EBV genome into any cell is possible, irrespective of whether it is normally infectable or not.
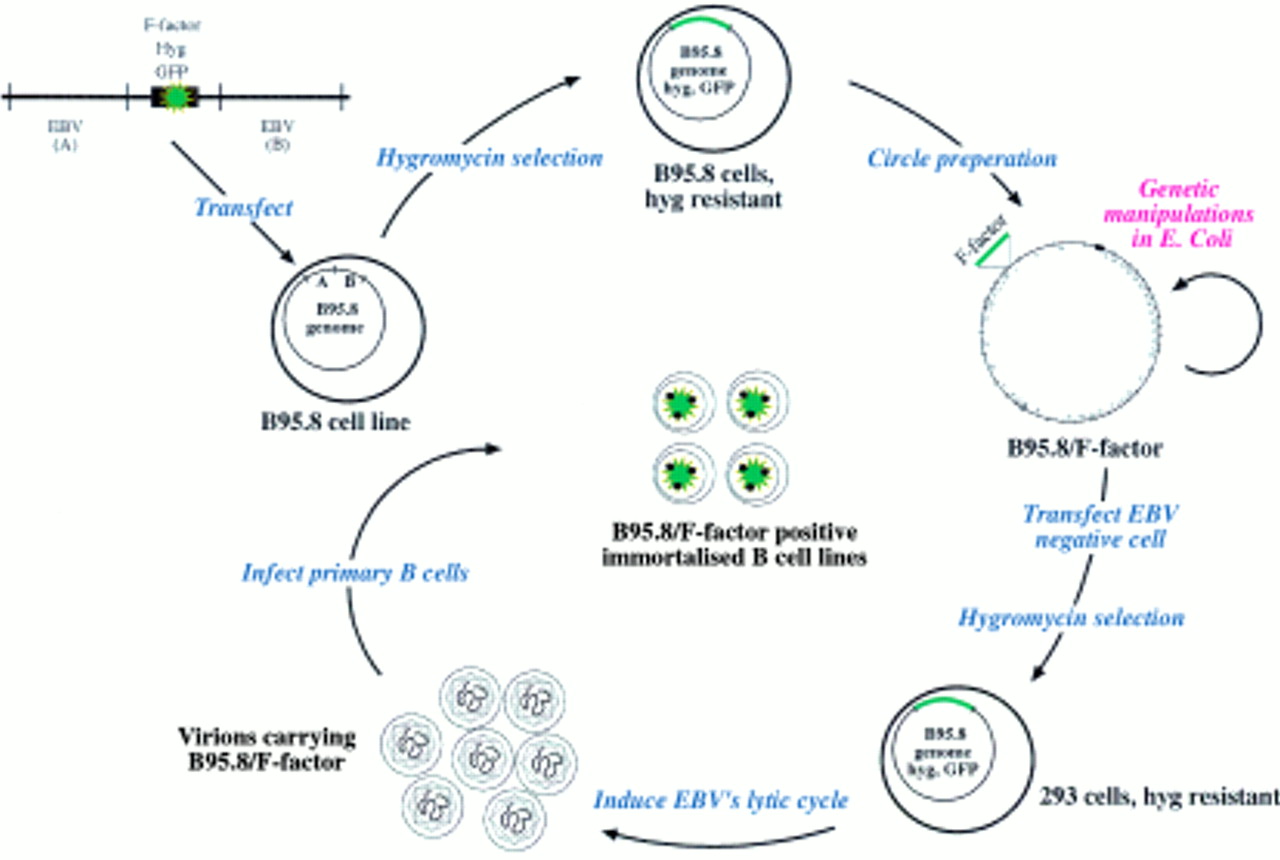
Epstein-Barr virus (EBV) genetic analysis in Escherichia coli: the maxi-EBV system. Schematic overview of the maxi-EBV system. A linearised DNA fragment consisting of an F-factor plasmid and two flanking regions A and B of EBV was transfected into the B95.8 cell line, which is latently infected with EBV. Homologous recombinations occur via the regions A and B to generate a B95.8 cointegrate, which encompasses the gene for hygromycin resistance (hyg) and green fluorescence protein (GFP), together with the F-factor replicon. Cells that contain such a cointegrate B95.8/F-factor survive under hygromycin selection. The preparation of circular DNA from these cells and its transfection into an appropriate E coli strain establishes the B95.8/F-factor molecule in E coli for further genetic modifications. The B95.8/F-factor DNA can be amplified and isolated from E coli in microgram quantities to be used for transfection into EBV negative cells, that is, 293 cells. Upon hygromycin selection, cell lines that carry the B95.8/F-factor molecule as extrachromosomal copies can be established. Induction of the lytic phase of the EBV life cycle yields viruses that carry the B95.8/F-factor molecule as genetic information. The infection of primary B cells leads to their immortalisation and B95.8/F-factor positive B cell lines.
Applications of the genetic analysis
DISSECTION OF THE VIRAL LATENT GENES AND THEIR FUNCTIONS
The construction of viral mutants in which a given gene has been inactivated provides important information about its function in the context of the complete viral genome. The early interest in the genetic analysis of EBV concentrated on those genes involved in the initiation and maintenance of B cell immortalisation. Most mutants generated and analysed so far are deletion mutants that allow the characterisation of genes and their products and whether they are dispensable or essential for the process of B cell immortalisation in vitro. In this review we will concentrate only on the description of EBV mutants and their phenotypes. For the discussion of the different genes and their functions we refer to two recent reviews that cover these aspects in detail.7,8
One of the most studied EBV genes, BNLF1 (its gene product is generally called latent membrane protein 1; LMP1), was found to be absolutely required for the immortalisation of primary B cells in vitro. LMP1 was also shown to be necessary for the continued proliferation and maintenance of the immortalised B cell in vitro.23–25 A further step in the dissection of the function of LMP1 was to generate viral mutants in which parts of the LMP1 protein were deleted. An EBV that carried a BNLF1 mutant allele in which the first 24 amino acids (aa) from the N-terminal cytoplasmic domain of LMP1 were deleted kept its ability to immortalise B cells.26 In contrast, the first 45 amino acids of the intracytoplasmic C-terminal part of LMP1 (aa 185–230) were found to be crucial for the function of LMP1, whereas the deletion of the remaining part of the C-terminus (aa 230–386) could be complemented by cultivating the primary B cells infected with the LMP1 mutant EBV on a feeder cell layer.27 The feeder cells were not absolutely required because a high virus titre could obviate the need for them.28 However, long term culture of the infected B cells was more efficient with the wild-type EBV, suggesting that the domain between aa 230 and 386 of LMP1 plays a crucial role in the long term maintenance of B cells in vitro.28 In fact, this LMP1 domain is likely to comprise additional subdomains with different functions because a virus carrying a BNLF1 gene with a deletion from aa 232 to 351 is not different to wild-type EBV with regard to B cell immortalisation.29 A more refined analysis of the domain comprising the first 45 amino acids of the intracytoplasmic C-terminal part of LMP1 provided indirect evidence for a crucial role of the tumour necrosis factor receptor associated factor (TRAF) binding site. Using overlapping cosmids Izumi et al constructed a viral recombinant in which the domain that engages the TRAFs was deleted (aa 185–211).30 After the infection of primary B cells with supernatants containing this viral mutant, as well as wild-type virus, none of 412 cell lines contained only the mutant virus, indicating that this particular domain of LMP1 is indispensable for B cell immortalisation.
The study of viruses in which the EBNA2 or EBNA3C genes were deleted also led to the conclusion that these gene products are absolutely required for B cell immortalisation in vitro,31,32 whereas EBNA3A seems to contribute to the initial process of B cell immortalisation only.17 In general, the genetic analysis of these nuclear proteins is less advanced than is the case for LMP1 or LMP2; however, at least four separate domains of the EBNA2 proteins were found to be essential for immortalisation, whereas disruption of additional domains diminished the efficiency of immortalisation.6,33–38 The EBNA-LP gene does contribute to the efficiency of B cell immortalisation6,31,39 and, as expected, EBNA1 seems to be essential for B cell immortalisation.40
The situation with LMP2A and LMP2B indicates that both LMP2 splicing variants do not contribute to the immortalisation process of mature resting B cells in vitro,41–46 although conflicting results have been published,47 and functional data argue for an essential role in the infection of differentiating B cells.48,49 Even more detailed mutations of LMP2A in the context of the complete EBV genome testify to the importance of LMP2A in the interference of immunoglobulin signal transduction.50–53
A virus that carries a deletion spanning the BARF0 gene kept its immortalising functions, suggesting that this 58 kb DNA fragment does not contain any essential immortalising genes.54 Similarly, the BHRF1, EBNA3B, BHLF1, BcRF1, BZLF1 genes, and the EBV encoded small RNAs (EBERs) do not seem to contribute to the immortalising potential of EBV in vitro.55–60
DISSECTION OF GENES INVOLVED IN THE EBV LYTIC PROGRAMME
Two transactivators, BZLF1 and BRLF1, are expressed very early during the EBV lytic cycle.61–63 The analysis of cell lines carrying EBV strains that cannot replicate, such as the Raji cell line, or genetically engineered strains in which the BZLF1 or the BRLF1 genes were disrupted, has shown that these proteins are required at all steps of the lytic cycle, including lytic DNA replication and the expression of late EBV genes.60,64 Some discrepancies were noted between the two studies, possibly because the Raji EBV genome carries multiple genetic abnormalities. A recombinant virus that carries a deletion of the BARF2 gene that encodes the single strand DNA binding protein was not able to support the lytic cycle, an observation in line with the function of this protein.12
The analysis of EBV virions in which the BHRF1, BHLF1, LMP2, or EBER genes were disrupted showed no difference compared with wild-type EBV in terms of lytic propagation.45,57,59,65 This observation suggests that the functions of these proteins during viral replication are redundant or are not apparent in vitro.
GENETIC ANALYSIS OF VIRAL INFECTION
The genetic analysis of genes involved in the process of cellular infection has just begun, although functional analyses so far have only provided limited information. EBV efficiently infects B cells but also certain other cell lines or primary cells that have epithelial characteristics. The BLLF1 gene encodes the viral glycoprotein gp350, which has been shown to mediate adhesion to target B cells.66,67 Surprisingly, infection of B cells or even epithelial cell lines was not abrogated with a BLLF1 negative EBV mutant, although the cells were infected with a lower efficiency.68 This finding implies the existence of additional viral ligands, as already observed in the case of herpes simplex virus 1 (HSV-1), another member of the herpesvirus family. In contrast, disruption of the BXLF2 gene, a viral gene that encodes a protein thought to mediate fusion between the viral and the cellular membranes suppressed the ability of EBV to infect its target cells16 (H-J Delecluse and W Hammerschmidt, 1999, unpublished data). A virus mutant that carries a mutation of the gp150 glycoprotein, encoded by the BDLF3 open reading frame, kept its ability to infect B cells and even showed an enhanced efficiency with regard to epithelial cell infection.69 The gB homologue of EBV, gp110, encoded by the BALF4 gene, is essential for virus formation,70,71 whereas the EBV glycoprotein gp42 encoded by the BZLF2 gene is crucial for viral infectivity of B cells but not of epithelial cells.72,73
CONSTRUCTION OF EBV STRAINS WITH NEW PROPERTIES
The potential of the technology used for the genetic analysis of herpesviruses is not limited to the study of the different viral functions but can be extended to design viral strains with properties of potential value in biotechnology.
A helper cell line for the encapsidation of EBV derived vectors
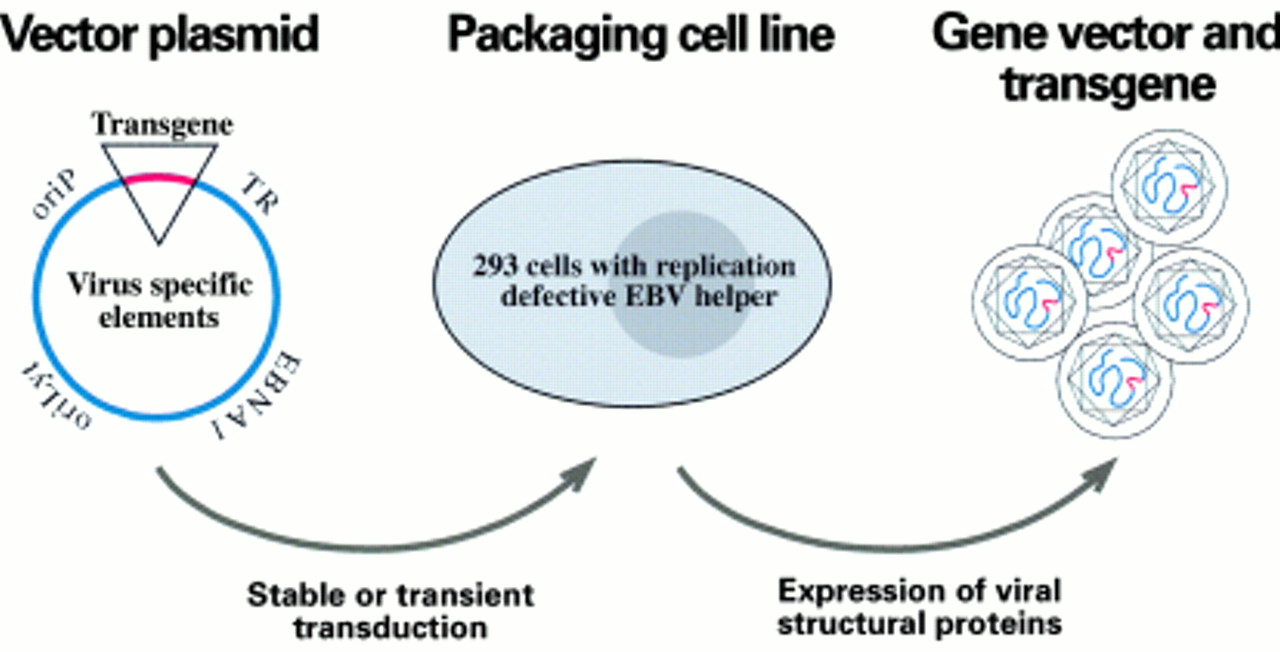
Most of the gene transfer systems based on viral vectors benefit from the use of a helper cell line that enables the packaging only of recombinant vector sequences. To accommodate foreign DNA fragments into viral vectors, viral sequences must be deleted from the viral genomes. In turn, the helper cell line provides the missing viral functions for encapsidation of the viral vectors. The helper cell line frequently contains a helper virus genome that provides all the necessary viral factors in trans to encapsidate the viral vectors, but lacks cis-acting elements, mostly packaging signals, to prevent its encapsidation into a viral particle (fig 6). Terminal repeats (TRs) are repetitive sequences of the EBV genome that are absolutely required for the incorporation of the viral DNA into the viral capsid.6,74 Because these sequence elements do not encode any protein essential for virus synthesis, a TR negative EBV strain provides a primitive but functional helper virus genome. Such a TR-less EBV genome was engineered in E coli, and a 293 cell clone in which this TR negative construct was introduced was able to encapsidate EBV derived vectors21 as anticipated.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Gene transfer system based on Epstein-Barr virus (EBV) derived vectors. Plasmids that carry the EBV replication origins (oriLyt and oriP), the EBV encoded nuclear protein 1 (EBNA1) gene, and the packaging sequences (TR) can accommodate large inserts as transgenes. These vectors are transfected into a helper cell line that carries a mutant EBV genome in which the TR was deleted. After completion of the lytic cycle, viral particles that carry the transgene are produced and can be used for the infection of target cells.
EBV based vectors of different sizes can be packaged in such a system as long as certain requirements are met. These include the presence of the cis-acting elements that allow DNA amplification and packaging in the helper cell line as well as extrachromosomal plasmid maintenance in the recipient cells. The gene vectors can deliver one or several genes of interest,21 or can be as complex as mini-EBV plasmids. In fact, mini-EBV plasmids themselves can be used to deliver genes of interest—for example, to manipulate autologous antigen presenting B cells for immunotherapy.75 The total packaging capacity of EBV based viral vectors is in the range of about 140 kb, which offers the opportunity to accommodate complete cellular genetic loci. With regard to the packaging viral genome, the flexibility to delete virtually all viral genes dispensable for virus synthesis will provide self limiting EBV helper virus genomes devoid of any potential oncogene, thereby preventing the accidental delivery of pathogenic genetic material in gene transfer settings.
Attenuated viral strains
The genetic analysis of EBV proteins expressed during latent infection led to the identification of viral genes that are essential for B cell immortalisation. Some of these genes found in in vitro immortalised B cells are also expressed in EBV associated tumours. Viral mutants that have lost one or more of these proteins are unable to immortalise B cells and can be considered as relatively harmless in terms of their pathogenic potential. These attenuated EBV strains could be used as live vaccines with the objective of preventing EBV associated diseases, such as the fatal mononucleosis that follows primary infection in patients with immunodeficiency. Because some of the latent proteins, such as LMP1, are frequently expressed in lymphomas, and in particular in the Reed-Sternberg cells of Hodgkin's disease (see KJ Flavell and PG Murray, this issue), it is likely that vaccination of the general population will reduce the incidence of these diseases. Vaccination trials using viral peptides from the gp350 viral glycoprotein have begun.76 Because immunity to EBV is mainly cellular in nature, the protection obtained by using attenuated live vaccines may be of higher quality.77,78 Systematic vaccination against a tumorigenic herpesvirus is common in the poultry industry and, as a consequence, the incidence of T cell lymphomas in chickens shortly after hatching, which follow infection with the Marek's disease virus, has diminished dramatically. In this case, vaccination is performed using live vaccine strains attenuated through multiple cell culture passages or herpesvirus isolates that normally infect turkeys.79 In fact, vaccination against Marek's disease virus has been the first successful attempt to prevent a natural virus induced tumour in a large animal population.
Conclusion and perspectives
Several experimental systems are now available that allow the manipulation of the genomes of large DNA viruses. The approach of cloning complete herpesvirus genomes has been used with herpesviruses, including mouse and human cytomegalovirus (CMV), herpes simplex virus, herpesvirus saimiri, and herpesvirus pseudorabies, and will be applicable to other γ-herpesviruses, such as the human Kaposi's sarcoma associated virus, HHV-8.80–84 These systems provide an unprecedented flexibility in the design of the recombinant herpesviruses. In the case of EBV, general interest is limited as long as the generation of immortalised B cell lines remains the only focus. Many EBV positive tumours in vivo differ greatly from immortalised B cell lines with regard to their viral gene expression pattern and frequently harbour additional genetic abnormalities such as chromosomal translocations. Therefore, more work on the cellular systems is required to exploit fully the potential of the genetic approach in investigating the pathogenesis of EBV associated diseases.
To date, viral vectors based on the EBV genome do not belong to the standard arsenal of vectors used in gene therapy trials. One reason for this situation is the difficulty in manipulating the EBV genome. It is likely that the recent availability of a helper cell line will allow the potential of these vectors for gene transfer to be evaluated, particularly in B cells. The large capacity of the EBV derived vectors, which allows insertion of large foreign DNA fragments, and the stability of EBV derived vectors in the infected host will certainly qualify these vectors for several applications.