Article Text

Abstract
Microarray technology allows the simultaneous analysis of up to thousands of different genes in histological or cytological specimens. Although microarray technology has so far mainly been applied in the research setting, its clinical application in pathology is expected in the foreseeable future. This paper presents an overview of the technical “ins and outs” of microarray technology, and discusses several putative applications in diagnostic pathology, which include tumour classification, the prediction of responses to certain chemotherapeutical or hormonal agents, the biological staging of tumours, the risk assessment of premalignant lesions, and the detection of microorganisms.
- microarray technology
- diagnostic pathology
- comparative genomic hybridisation
- gene sequencing
- gene expression
Statistics from Altmetric.com
- microarray technology
- diagnostic pathology
- comparative genomic hybridisation
- gene sequencing
- gene expression
The morphological analysis of the phenotypic characteristics of diseased tissue is still the basis of diagnostic pathology. In many pathology laboratories additional information is, on a routine basis, obtained by immunohistochemistry,1 quantitative measurements,2 or electron microscopy. Owing to the rapid developments in molecular biology in recent years, many clinicians and basic researchers in the field of oncology are focusing on the analysis of the genetic alterations and gene expression of tumours, as well as premalignant lesions, which underlie their biological properties and probably their clinical behaviour. This type of molecular analysis can be useful for diagnosis making and prognosis assessment in different diseases.3 This is particularly true for tumours because a correct (sub)classification of cancer is essential for guiding treatment—for example, deciding between surgery, radiotherapy, chemotherapy, immunotherapy, or combined approaches. Gene expression patterns of tumours can often tell the oncologist in advance whether a patient will respond to certain chemotherapeutical or hormonal agents.4 With this information, precious time can be won in the treatment of these patients, and ineffective treatments avoided. Other examples are found in the areas of the pathological classification of tumours, biological staging, and risk assessment of pre-malignant lesions. Many of the gene expression profiles or genetic alterations correlate with the patients' prognoses and can guide clinical follow up strategies. In addition, data on genomic changes and altered gene expression patterns associated with different steps in tumour progression increase our knowledge of tumour development and open up new possibilities for cancer prevention, such as a more rationalised design of health care screening programmes and the development of novel chemopreventive agents.
For these reasons, there is a huge interest in the monitoring of (global) gene expression and the genetic alterations of tumours. Until now, laboratory techniques were not suitable for the measurement of the expression of large numbers of genes at the mRNA level (for example, northern blotting, the reverse transcriptase polymerase chain reaction (PCR), and in situ hybridisation) or at the protein level (western blotting and immunohistochemistry). One has to realise that, on average, 10% of the estimated 100 000 genes present in the human genome are expressed in a given cell. Similarly, for the measurement of genetic alterations, the choice was between high resolution but with a limited overview (for example, changes in single bases evaluated by mutation analysis, Southern blot hybridisation, direct sequencing, and in situ hybridisation), or a global overview with a limited resolution (for example, analysis of changes at the chromosomal level by comparative genomic hybridisation5 or karyotyping of cultured cells). Furthermore, all these techniques are relatively complex and require special equipment and expertise that are not available in many laboratories. Moreover, these techniques are labour intensive and therefore expensive. These drawbacks have hampered the introduction of these useful techniques in diagnostic pathology.
Recently, a microarray technology has been developed that might overcome many of these drawbacks. It increases the possibilities both for the analysis of gene expression and the monitoring of genetic changes. It provides the best of both worlds: it is not labourious, has high resolution, and a global overview. A microarray is a series of DNA target sequences spotted on to a carrier (a glass slide, a silica “chip”, or a membrane) in a logical and orderly fashion, on which nucleic acid probes derived from (for example) a tumour are hybridised. Four types of microarrays can be discerned and each has different applications (table 1): (1) arrays of human genomic DNA, (2) arrays of human cDNA, (3) arrays of human oligonucleotides, and (4) arrays of bacterial or viral DNA or cDNA.
Overview of the four types of microarray
Although, to date, microarray technology has been applied mainly in research settings, it is developing so rapidly that it will probably soon find its place in the diagnostic pathology laboratory. Here, we present an overview of the technical “ins and outs” of the different microarray techniques, and will try to explain how this technique might be useful in diagnostic pathology to help the pathologist in making diagnostic and/or prognostic decisions.
Microarray comparative genomic hybridisation (CGH)
In CGH, genomic gains and losses can be analysed “genome wide” in a single experiment by comparing tumour DNA and normal reference DNA. There is no need for cell culturing and the DNA can be isolated from any source. In classic CGH, tumour DNA (detected in green) and normal DNA (detected in red) are labelled by nick translation and hybridised simultaneously on to normal metaphase chromosome preparations.5,16 Using fluorescence microscopy and digital image processing, the green to red ratios along the axis of each individual chromosome can be measured. As a result of the use of metaphase chromosomes, the resolution is limited to 10–20 Mb. This limitation makes it difficult to link copy number changes to the genes involved. In addition, the need to perform karyotyping in every experiment to identify the targets limits a widespread application of this technique.17–19
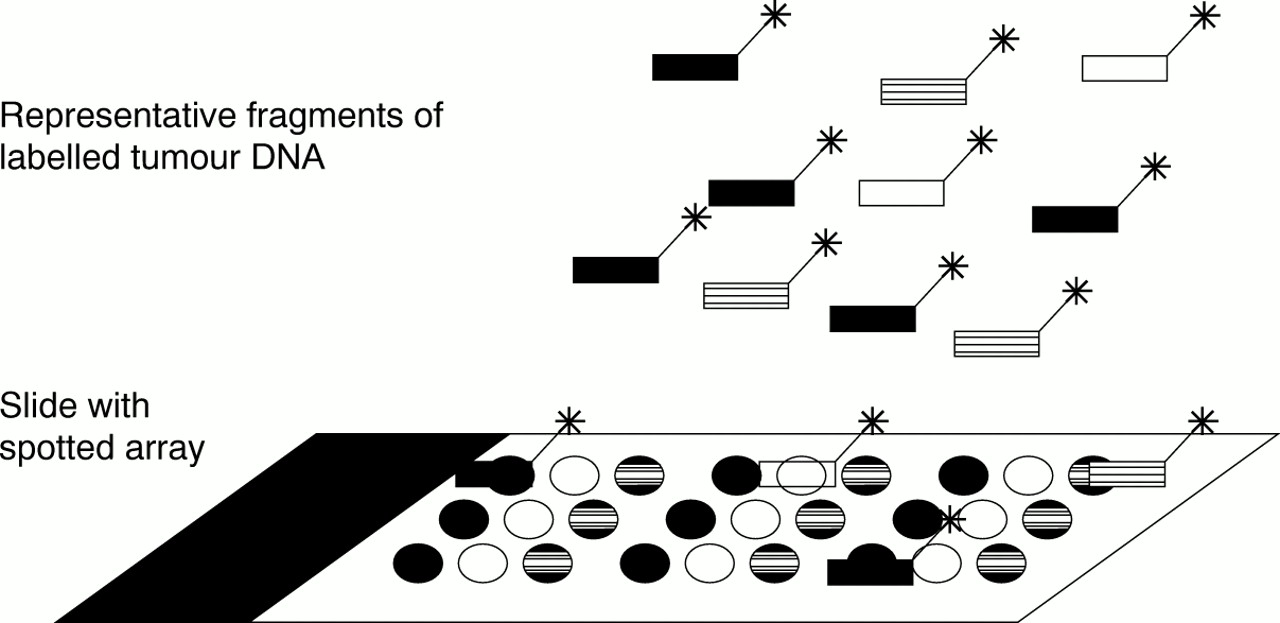
In microarray CGH, the substrate is not a normal metaphase spread, but an array of DNA fragments (100 bp to 100 kb), and the precise chromosomal locus of each is known (fig 1). In this way, by using an array of approximately 5000 spots, a genome wide analysis for gains and losses at a 1 Mb resolution is possible.6
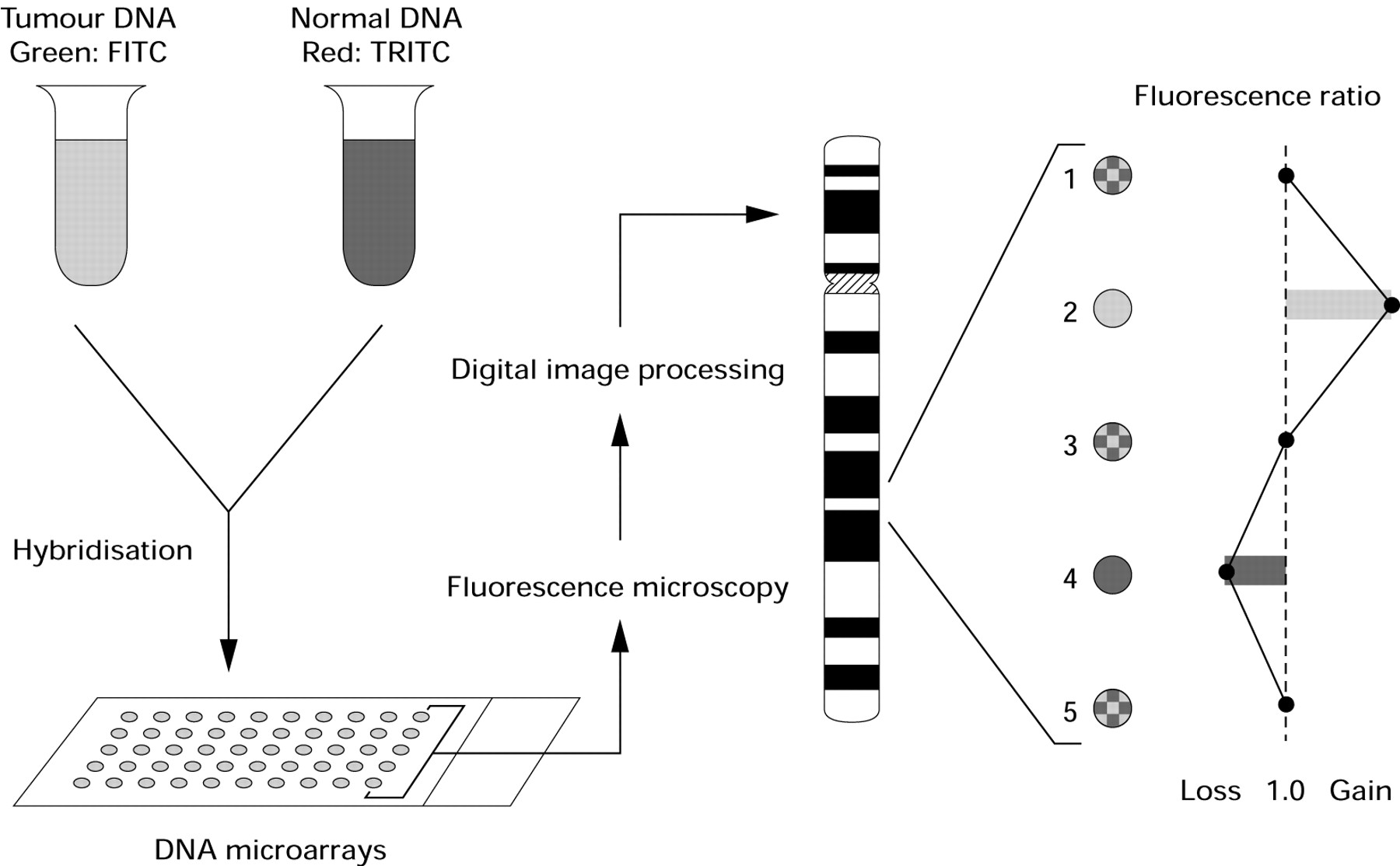
Principle of microarray comparative genomic hybridisation. FITC, fluorescein isothiocyanate; TRITC, tetramethyl rhodamine isothiocyanate.
Like conventional CGH, microarray CGH can be useful for several purposes where more simple techniques such as immunohistochemistry fail. In the case of a second tumour in a patient (such as a lung cancer after a larynx cancer), patterns of gains and losses, as established at high resolution by microarray CGH, can determine whether the tumour is a metastasis of the first tumour (similar patterns will be seen) or a second primary (different patterns will be seen). Similarly, in the case of a metastasis in a patient with two (or more) primary tumours, the origin of the metastasis can be traced, which might be important for future treatment. In addition, patterns of gains and losses might prove to be useful for the classification of tumours, a risk assessment of premalignant lesions, and the prognosis prediction of cancers.
Microarray expression analysis
In microarray expression analysis, the focus is not on differences in DNA copy numbers between normal and (for example) tumour tissue, but on differences in gene expression levels between normal and abnormal tissue (for example, tumours, inflammatory and degenerative diseases). This approach allows the simultaneous analysis of the expression of thousands of genes in a single experiment. Arrays of cDNAs of known (or for research purposes even unknown) genes are spotted on to a carrier (a glass slide, a nylon filter, or a silica chip). Subsequently, labelled RNA or cDNA probes synthesised from mRNA isolated both from abnormal and normal tissue are co-hybridised on to the array. The signal intensities for every spot can be determined, which provides a measure of the expression of thousands of genes in a single experiment.20 This technique does not only yield information about the expression of individual genes in different tissues, but also on the regulation of different genes organised in pathways.
Microarray expression analysis could also be useful for several purposes in clinical practice. Similar to microarray CGH, in the case of a second tumour in a patient (for example, lung cancer after a larynx cancer), expression patterns establish whether it is a metastasis of the first tumour (similar patterns will be produced) or a second primary (different patterns will be produced). Similarly, in the case of a metastasis in a patient with two (or more) primary tumours, the origin of the metastasis can be traced, which is of course important for future treatment. In addition, expression patterns could be useful for the classification of tumours21 and a risk assessment of premalignant lesions. Other potential applications concern monitoring the response of tumours to treatment, an assessment of prognosis, and a prediction of drug resistance patterns of certain diseases.
Mutation and/or polymorphism analysis
An even more sensitive approach is one in which microarrays allow the detection of changes at the level of individual nucleotides; that is, mutations or polymorphisms. In this type of microarray, probe DNA is non-competitively hybridised to an array of specific target DNA sequences that differ only in specific nucleotides. In this way, p53 mutations (for example) can be detected.22 In addition, polymorphisms on thousands of genes can be analysed in a single experiment.23
Potential or already established applications of this technique concern gene sequencing, such as for the p53 and BRCA1/2 genes, and in dystrophinopathies. Using polymorphism analysis, risk profiles could be established for the development of certain diseases, such a chronic inflammatory diseases, degenerative diseases, cardiovascular diseases, and tumours.
Microorganism analysis
For the detection and classification of microorganisms, the main goal is to detect the presence of nucleic acid sequences specific to these microorganisms by hybridising probe DNA non-competitively to arrays of bacterial or viral DNA or RNA of known origin. A well known example is the human immunodeficiency virus (HIV) chip,24 and human papillomavirus (HPV) typing could be another important application.
Details of the technique
Microarray technology is currently commercially available, but very expensive. In addition to the high costs, these commercially available microarray systems (for example, Micromax, NEN Life Science Products, http://www.nenlifesci.com or GeneChip, Affymetrix, http://www.affymetrix.com) are not flexible enough for changes in the experimental setting, and it would be very difficult (if possible at all) to obtain and scan customised arrays. In contrast, other systems are available that allow the preparation of arrays with varying numbers of clones of choice, independent of supplying companies. These systems are suitable for cDNA as well as genome clone arraying and allow the use of small probe samples, therefore saving crucial clinical material. This paper will focus on systems that use these laboratory made microarrays.
WETWARE
An important issue is the availability and handling of libraries with cloned and amplified cDNA or genomic DNA. For small scale use, target DNA sequences can be derived from public databases in the public domain and the clones obtained by PCR and molecular DNA cloning. However, for genome wide screens large libraries need to be available. A sequence confirmed human expression (cDNA) library for genome wide screens is commercially available (Research Genetics, http://www.resgen.com) (currently 40 000 clones). However, it should be realised that such a library needs to be updated periodically and that its use and maintenance is extremely labour intensive.
For microarray CGH, a genome wide library of DNA clones that covers all chromosomes at 1–3 Mb resolution is desirable, supplemented with clones for regions of particular interest for cancer research. This would mean a set of approximately 3000–5000 clones. Initiatives in this field are under way, but these types of libraries are not yet available. Alternatively, clones can be selected from public domain libraries as bacterial artificial chromosomes (BACs; 100 kb; 30 000 clones), yeast artificial chromosomes (YACs; 1 Mb; 3000 clones), cosmids (40 kb; 75 000 clones), and phages (20 kb; 150 000 clones).
MICROARRAY MANUFACTURING
Initially, pioneers in the field used laboratory made equipment as designed and published by Brown and co-workers20 (webpage of PO Brown: http://cmgm.stanford.edu/pbrown). Recently, several commercial array systems have been developed for printing target DNA on microscope slides (“gridding”). In short, every arrayer consists of an XYZ robot of which the printhead travels between a microtitre plate, containing the target DNA, and the microscopic glass slide (fig 2), printing approximately 2500 spots/cm2 when target spots are on 250 μm centres. The differences between the available systems are mainly based on the structure of the printhead: there are several different ways of picking up a small amount of target DNA solution and printing small amounts of this solution in an orderly and systematic way on a microscope slide. Examples of gridding methodologies include piezo technology (inkjet dispensers), quills, and “pen and ring” systems. As a consequence, the systems differ considerably in reliability, accuracy, capacity, and the required (starting) volume of sample.

Principle of the gridding or arraying machine.
TEST SAMPLE OR PROBE PREPARATION
An important element in the technology is the amount of uncontaminated test DNA or mRNA that is used as a probe. In particular, premalignant lesions are often small, and frequently biopsy samples only are available. Another major problem is the contamination of tumour tissue with stroma and inflammatory cells. This type of tissue contamination “dilutes” the tumour sample and reduces the differences found between the tumour and the reference sample, giving rise to aberrant data points. To keep contamination at a minimum, microdissection is often desirable.25 To deal with both problems, different studies are being conducted at the moment, aimed at an amplification method for RNA as well as DNA, which should be representational as well as linear (fig 3). When these procedures are available, they will have a major impact on the possible applications of microarray technology in the diagnostic setting. Only one or a few (tumour) cells would then be needed for the analysis of expression patterns and genomic abnormalities. After isolation, and possibly amplification, test DNA and RNA always need to be labelled with a fluorescent dye according to standard protocols before use as a probe.
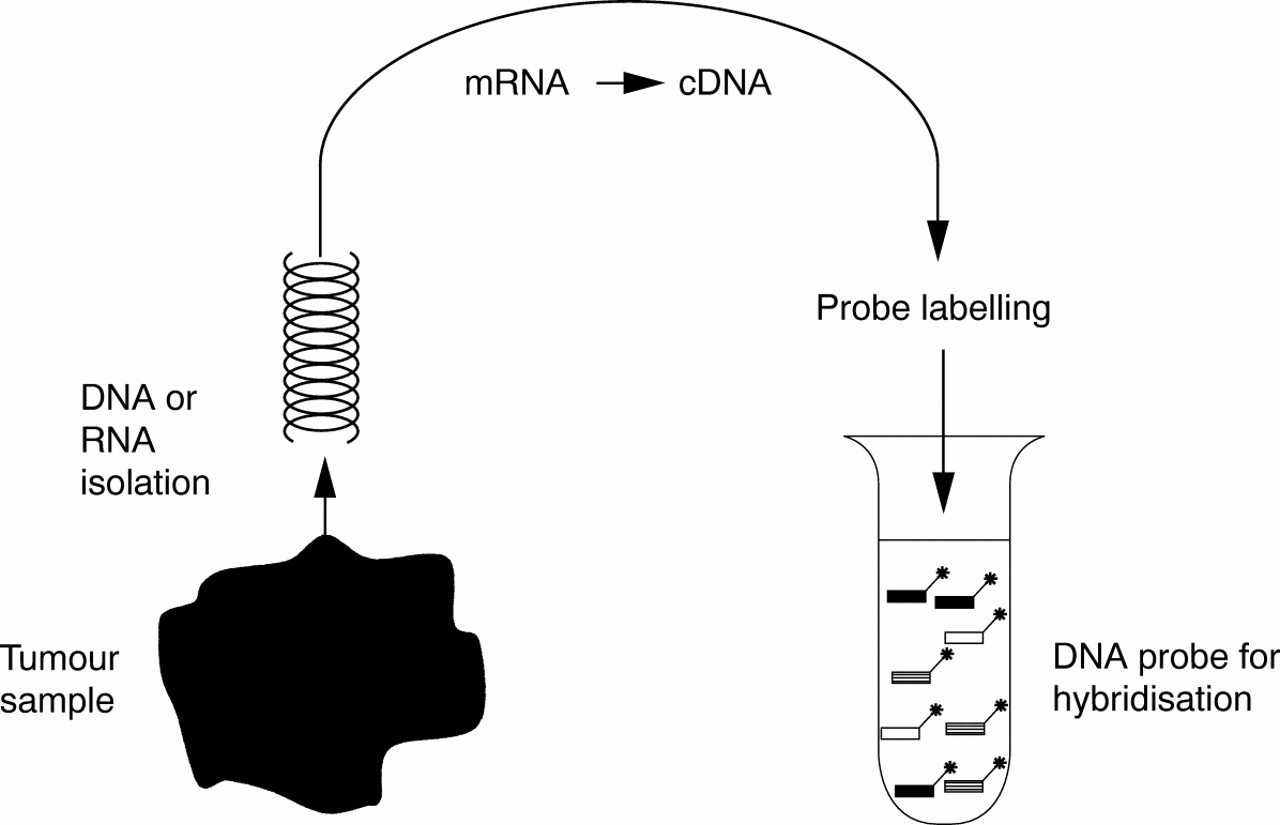
Representational probe preparation.
HYBRIDISATION
For microarray CGH and expression analysis, probes derived both from reference and tumour tissue are hybridised simultaneously to one microarray (fig 4). By using different fluorescent labels for each of the probes, the microarray can be scanned separately for the tumour and reference tissue sample. By comparing the results, differences in genomic or gene expression patterns between the tumour and the reference tissue can be detected.
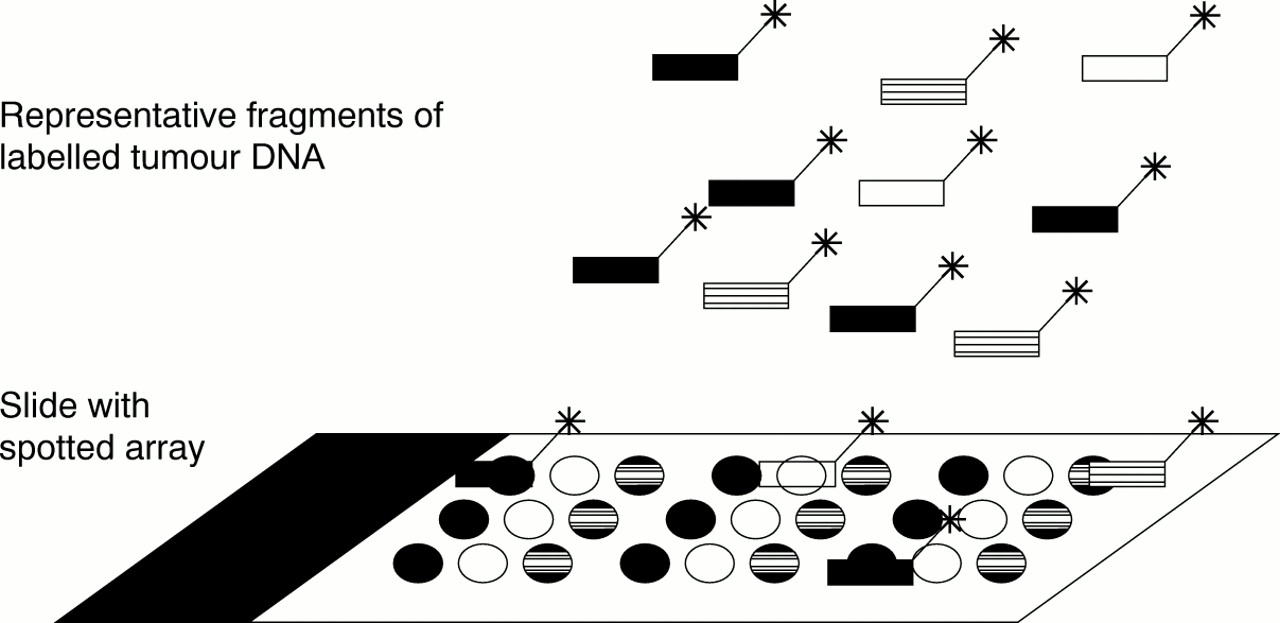
Hybridisation of labelled probes to spotted array.
SCANNING THE ARRAY
After hybridisation, a digital image of the array is made by a scanner. Most scanners are either confocal laser or standard fluorescent microscope based, further equipped with a filter wheel and a CCD camera (wide field imaging systems). Laser scanners are less sensitive to background staining in comparison with wide field imaging systems, because only the light emission from the target spots is detected. However, fluorochromes differ in their excitation spectra, and several lasers may be required. In practice, the combination of 532 nm (green) and 633 nm (red) lasers with Cy3 and Cy5 as fluorochromes is used most frequently. However, other fluorochromes might be easier to incorporate into the probe and may give a more stable and acceptable result, although they might require other lasers.
A wide field imaging system is based on a CCD camera that captures the array, or part of the array, and projects it on to a CCD chip using a mercury lamp instead of scanning the array with a laser beam. Such a system can sometimes be less expensive than a confocal laser scanning microscope, and more flexible as to the choice of fluorochromes because only a different filter has to be installed. However, in contrast to the confocal laser scan microscope, the wide field imaging system is influenced by background noise, which may disturb the calculated fluorescent ratios (see below).
IMAGE ANALYSIS AND DATA HANDLING
Once a digital image of the array is obtained, it must be analysed. Because image analysis on films or phosphor image representations of radioactive hybridisations is difficult, we will focus on the image analysis of fluorescence signals obtained from robotic printed glass slides. Robotic printing results in a highly regularly shaped and spaced microarray for which modern image segmentation methods, such as mathematical morphology, can be used to predict the likely shape and placement of the hybridisation signal. Because of the regular spacing, corrections for background and uneven illumination can be performed locally. Artefacts are easily detected (irregular shape or displacement of spots) so that they can be removed. For each segmented spot, the red to green fluorescence ratio is then calculated. These ratios are then translated into a table containing the ratios of intensities of every individual spot on the array (webpage of Eisen MB: http://rana.stanford.edu/). Subsequently, specific ratios can be allocated to specific clones; that is, genes (fig 5). Because high density arrays produce an extremely large amount of information, databases are required for the evaluation of information obtained after each experiment. With respect to handling and analysing these large amounts of data, terms such as “data mining” and “bio-informatics” have been introduced.26 This type of analysis demands reliable, rapid, and user friendly software and hardware, combined with a high storage capacity. The biological meaning of the data can be investigated by using complex computational analysis, such as clustering of co-expressed, co-gained, or co-lost genes.27

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Computer based image analysis and the generation of a table with gene expression data (ratios).
Future perspectives
Microarray technology is still in its infancy; there are still more people discussing the technique than using it. There are several reasons for this: the high costs, the need for specialised technical expertise, the need for collaboration between different disciplines, the problem of coping with very large amounts of data, and the uncertainty about the biological meaning and clinical relevance of the results.
Notwithstanding the fact that not all of these elements have been solved, it should be realised that the application of microarray technology has the possibility to change the practice of diagnostic pathology dramatically, probably even more than the introduction of immunohistochemistry. Not only might the results of microarray analysis one day prevail over histopathological diagnoses with respect to the guidance of treatment and the assessment of prognosis, but it is also possible that, in the near future, histopathological entities will be redefined on the basis of genomic and gene expression patterns at a much higher rate than is happening already. Especially in fields where diagnostic categories overlap and form a spectrum, as in soft tissue tumours, such a redefinition of entities is likely to occur.28 If pathologists neglect the potential of these processes at an early stage, it is not unthinkable that in the future their task will be limited to judging whether or not a sample contains adequate material for microarray analysis or whether a tumour's histology is compatible with the more precise diagnosis made by geneticists or chemists, or just handing over samples to specialists from other disciplines after dissection. Even worse, such samples might be taken from the specimen before they are sent to the pathology department. This latter situation is not only a worst case scenario for pathology as a profession, but it may also be harmful to the patient, because adequate sampling of representative tissue for diagnostic microarray analysis certainly requires the expertise of a trained pathologist.
However, there is no need to be pessimistic yet, because when pathologists embrace this promising new technique, it may not only make their work easier and more interesting, but it might also provide valuable scientific information that can help to fill gaps in our knowledge of many diseases.