Article Text

Abstract
Aims—To establish whether the ectopic expression of CCN3 (NOV) in glioma cells can interfere with their tumorigenic potential and assess its potential value in molecular medicine.
Methods—Glioma cell lines were used to assess whether differences in the degree of intracellular communication induced by the expression of the gap junction protein connexin 43 (Cx43) is related to the differential expression of CCN3 (NOV). The antiproliferative activity of rat CCN3 (rCCN3; NOV) in glioma cells, has been assessed both in vitro and in vivo with glioma cell lines expressing different amounts of CCN3 (NOV).
Results—Upon ectopic expression of Cx43, the growth of C6 glioma cells is decreased. An increase of CCN3 (NOV) expression matches the reduced tumorigenic potential of these transfected cells. The localisation of CCN3 (NOV) is affected by the increased expression of Cx43 in the Cx-13 transfected cells, in which it is detected at areas of cell–cell contact. In a xenograft model, CCN3 (NOV) transfected glioma cells were found to induce tumours to a lesser degree than their parental counterparts, which do not express detectable amounts of CCN3 (NOV).
Conclusions—Previous observations had suggested an inverse relation between CCN3 (NOV) expression in glioma cells and their tumorigenicity. These results establish a direct association between the establishment of functional gap junctional intercellular communication and the expression of rCCN3 (NOV). In addition to a negative effect on murine and human cell growth, CCN3 (NOV) has antiproliferative activity on tumour cells in vivo. Thus, the antiproliferative activity of the CCN3 (NOV) protein might involve reorganisation of cellular contacts that play a crucial role in tumorigenesis. The antiproliferative activity of CCN3 (NOV) established in this work sets the stage for the potential use of CCN proteins in molecular oncology.
- NOVH
- CCN3
- CYR61
- CTGF
- human glioma
- extracellular matrix
- connexin 43
- gap junctional intercellular communication
Statistics from Altmetric.com
- NOVH
- CCN3
- CYR61
- CTGF
- human glioma
- extracellular matrix
- connexin 43
- gap junctional intercellular communication
The CCN3 (NOV) gene1 was originally isolated as a target site for myeloblastosis associated virus in avian nephroblastoma, which is a unique animal model of the Wilms's tumour.2 It is a member of the CCN family of genes,3 which encode multimodular regulatory secreted proteins reported to play roles in fundamental biological processes, such as cell attachment and proliferation, differentiation, angiogenesis, and development.4–6
In addition to the three prototypic founding members (CYR61 (cysteine rich), CTGF (connective tissue growth factor), and NOV (nephroblastoma overexpressed)), three other CCN proteins (WISP1–3 (Wnt induced secreted proteins)) have also been described in humans.7–10 Further to a recent nomenclature proposal (First International Workshop on the CCN Family of Genes), these proteins are now designated under the CCN acronym, with numerals corresponding to the chronology of their discovery. Four structural modules sharing partial identity with insulin-like growth factor binding proteins, the Von Willebrand factor type C repeat, thrombospondin type 1 repeat, and the C-terminal portion of several extracellular growth factors have been recognised in the CCN proteins.3 Despite their similar multimodular architecture and conservation of 38 cysteines, the CCN proteins exhibit distinctive biological properties,4–6,11 which might result from different combinatorial events.6 Therefore, it appears that the CCN proteins are capable of stimulating or inhibiting cellular growth in normal conditions, depending upon their cellular localisation and time of expression.6
The human CCN3 (hCCN3; NOV) protein is widely detected in the developing human embryo,12 with an expression pattern partially overlapping that of other CCN family members.4–6 The association of hCCN3 (NOV) with the extracellular matrix,12,13 and the interaction of hCCN3 (NOV) with fibulin 1C,14 both suggest that hCCN3 (NOV) plays a role in cell attachment signalling. The induction of cell quiescence15 and cell growth arrest1 by chicken CCN3 (NOV) and the stimulatory effects of hCCN3 (NOV) on NIH3T3 cell growth16 account for hCCN3 (NOV) expression both in terminally differentiating and actively proliferating cells.
Because hCCN3 (NOV) expression is tightly correlated with the development of the human nervous system,17 and hCCN3 (NOV) protein is detected along neuronal axon processes within human kidney,18 we questioned whether abnormal expression of this protein might be associated with brain tumour biology. Our previous observations indicated that glioma and astrocytoma cells that expressed the highest amounts of CCN3 (NOV) were those exhibiting the least aggressive phenotype,19 suggesting an inverse relation between tumorigenicity of these tumour cells and CCN3 (NOV) expression, a conclusion consistent with the antiproliferative properties of CCN3 (NOV).
In our present study, we examined whether the expression of the rat CCN3 (rCCN3; NOV) correlated with the degree of cell–cell communication induced by the gap junction protein connexin 43 (Cx43), and investigated the antiproliferative activity of hCCN3 (NOV) in glioma cells, both in vitro and in vivo.
Materials and methods
DIFFERENTIAL DISPLAY AND SEQUENCING
To identify differentially expressed RNA species, we used the Genomyx Hieroglyph mRNA profile system (Genomyx Corp, San Francisco, California, USA) and the GenomyxLRTM DNA sequencer. A 200 ng sample of total RNA was treated with DNAse I (Gibco, Cergy Pontoise, France), and the reverse transcription was performed with 10 different 3′ primers consisting of T7 partial sequence followed by T12 and two anchored bases in different combinations, according to recommended procedures. Reverse transcribed duplicate RNA samples were subjected to polymerase chain reaction (PCR) amplifications using the same anchored 3′ primer and different 5′ arbitrary primers and using cold dNTPs, 33P-dATP, AmpliTaq DNA polymerase, and PCR buffer containing 15mM MgCl2. The PCR products were separated on a 4.5% urea polyacrylamide gel, which was exposed to x ray film and differentially displayed bands selected and excised from the gels. These samples were re-amplified with M13 reverse (24 mer) and T7 primers in a secondary PCR reaction using AmpliTaq gold (Perkin Elmer Cetus, Foster City, California, USA). Some of the resulting products were separated on low melting point agarose gels to produce radiolabelled probes for northern blot analysis, and the remaining reamplified products were treated with alkaline phosphatase and exonuclease I and then bidirectionally sequenced.
ANTIBODIES
CNBrSepharose (Pharmacia, San Quentin en Yvelines, France) was swollen in 10 ml 1mM HCl and washed with 200 ml of the same solution before transfer into 0.8 × 4 cm Poly-Prep chromatography columns (BioRad, Marnes la Coquette, France). The volume of the gel in the column was routinely 3.5 ml. A total of 3.8 mg K19M peptide18 was dissolved in 5 ml 0.1M NaHCO3 containing 0.5M NaCl and mixed with the CNBr gel within the column, by gentle rotation on a spinning wheel for two hours at room temperature.
The column was washed five times with 3 ml of 0.1M NaHCO3 containing 0.5M NaCl and with 3 ml of 0.1M Tris/HCl (pH 8.0). After resuspending the gel in 5 ml of 0.1M Tris/HCl (pH 8.0), the column was rotated for 90 minutes at room temperature and washed three times with 5 ml of 0.1M acetate buffer (pH 4.0), 0.5M NaCl; 5 ml of 0.1M Tris/HCl (pH 8.0), 0.5M NaCl; and 3 ml phosphate buffered saline (PBS; pH 7.4).
For purification of the anti-hCCN3 (NOV) antibodies, a 2 ml sample of anti-K19M antiserum (diluted with 1 ml PBS) was used to resuspend the gel and incubated on the rotating wheel for one hour at room temperature. After collecting the flow through, the column was washed twice with 40 ml of PBS. The bound antibodies were eluted with 5 × 3 ml of 0.2M glycine (pH 2.7) and collected in tubes containing 0.3 ml 1M Tris (pH 11.0).
For desalting, the 3 ml samples of each bound fraction were loaded on to a PD-10 column (Pharmacia). Once the flow through was discarded, the proteins were eluted with 3.5 ml of 0.1M phosphate buffer (pH 7.0). Purification of IgGs was performed on a HiTrap protein A column (Pharmacia). After washing the column five times with 1 ml of 0.1M phosphate buffer (pH 7.0), the bound fractions (K19M-AF-BFs) were loaded as 1 ml samples and the column was washed with 5× 1 ml PBS. The bound IgGs were eluted with 5 × 1 ml 0.1M citric acid (pH 3.0) in test tubes containing 0.3 ml 1M Tris (pH 8.0), and their specificity was checked by western blotting.20
Rabbit anti-rCCN3 (NOV) polyclonal antibodies have been described previously.21
IMMUNOBLOTTING OF CCN3 (NOV) CONTAINED IN CONDITIONED CELL CULTURE MEDIUM
For hCCN3 (NOV) detection, 1 ml fractions of conditioned media were collected from P60 plates with cells growing in monolayer and mixed with 50 μl of a 50% heparin sepharose slurry in PBS. After an overnight incubation at 4°C on a rotating wheel the sepharose beads were centrifuged at 23 000 ×g for 30 seconds, boiled for 10 minutes in sample buffer, and the dissolved proteins were electrophoresed on 15% Tris/HCl precast gels (BioRad), before transfer on to PVDF membranes (Millipore, San Quentin en Yvelines, France). Blots were incubated with affinity purified K19 anti-CCN3 (NOV) polyclonal antibody (1/500) for one hour in standard buffer (1× Tris buffered saline (TBS), 5% non-fat dry milk, 0.1% Tween 20). After washing, the blots were incubated with horseradish peroxidase labelled goat antirabbit IgG (1/5000) (Biorad) for one hour, and detected by enhanced Chemiluminescence (Amersham, les Ulis, France).
CELL CULTURE
The G59 cells were originally isolated and provided by Dr M Westphal. The G59W540 cells expressing hCCN3 (NOV) were isolated in B Perbal's laboratory. They were stably transfected by means of the pCMV82nov vector.18 The rat C6 and human U87-MG glioma cells were cultured under standard conditions. All cells were maintained at 37°C in 5% CO2 using DMEM medium supplemented with 10% fetal calf serum and 1% streptomycin and penicillin. Growth curves were performed by plating 5 × 104 cells into tissue culture plates and counting cells with a haemacytometer at the indicated days. The Cx43-13 C6 transfected cells were obtained and maintained as described previously.22
ANIMAL STUDIES
G59W and G59W540 xenografts were grown in the hindlimbs of 6–8 week old athymic female nude mice (Frederick Cancer Research Institute, Frederick, Maryland, USA) by subcutaneous inoculation of 5 × 106 cells suspended in 100 ml PBS. Tumours were allowed to grow for 11–15 days to a mean (SD) volume of 470.3 (28.1) mm3 (n = 51). At day 0, initial tumour volume was determined by direct measurement using calipers. Subsequently, tumour volume was determined two to three times each week. Based on the day 0 tumour volume, mice were randomly assigned to treatment groups such that the mean volume of each treatment group was approximately equal. The care and treatment of experimental animals was in accordance with institutional guidelines. Data are reported as per cent of original (day 0) tumour volume and plotted as fractional tumour volume (SEM).
IMMUNOHISTOCHEMISTRY OF VESSELS
Xenografts were surgically resected, snap frozen in OCT compound, and 4 μm sections were cut and mounted on poly-L-lysine coated slides. Sections were fixed in 1% paraformaldehyde (MeOH free) (Polysciences, Warrington, Pennsylvania, USA). After blocking with goat serum, and quenching endogenous peroxidase activity, slides were incubated at 4°C overnight with rat antimouse CD31 monoclonal antibody (Pharmingen, San Diego, California, USA), titred to 0.1 μg/ml. The sections were incubated for 25 minutes at 37°C with rabbit antirat IgG biotinylated secondary antibody (2 μg/ml) (Vector, Burlingame, California, USA), which had been preabsorbed with 1% normal human serum (Jackson ImmunoResearch, West Grove, Pennsylvania, USA). Appropriate negative controls were performed with rat IgG2a isotype control (Pharmingen), CD31 monoclonal antibody alone, and secondary antibody alone. ABC Vectastain Standard Elite (Vector) was applied and the sections were developed with the Vector DAB kit. Each slide was counterstained with a solution (0.03%) of light green SF yellowish (Fisher, Itasca, Illinois, USA). One investigator who was blinded to the treatment groups read the slides. Based on the size of the tumour section, three non-necrotic, high power fields (×400) at the tumour periphery were examined using the Nikon Microphot-FX microscope equipped with a Sony digital camera. Vessels were counted using Macintosh Image Pro-Plus software. The mean vessel count was determined for each tumour section.
HISTOPATHOLOGY OF NECROSIS
Sections (4 μm) were cut from OCT blocks and mounted on poly-L-lysine coated slides. Sections were fixed in 1% paraformaldehyde (MeOH free), stained with haematoxylin and eosin, and examined for total necrosis/tissue section. One investigator who was blinded to the treatment groups read the slides. The entire tumour section was scanned (×40) and three high power fields were examined for necrosis. The mean per cent necrosis was determined for each tumour section.
Results
The expression of rCCN3 (NOV) is induced upon gap junctional coupling in rat glioma cells. Rat C6 glioma cells show both decreased concentrations of the gap junction protein Cx43 and limited intercellular communication.23 There is evidence to suggest that reduced cell–cell communication by gap junctions found in cancer cells accounts, in part, for their tumour characteristics.24 Several studies have linked aberrant gap junctional intercellular communication (GJIC) in tumours, downregulation of GJIC by cancer causing agents or genes, and upregulation of GJIC by inhibitors of carcinogenesis.24 In addition, the introduction and overexpression of connexin cDNAs in tumour cells suppresses the growth and/or tumorigenicity of many transformed cells. We have shown that transfection and expression of Cx43 in C6 glioma cells suppresses growth.22
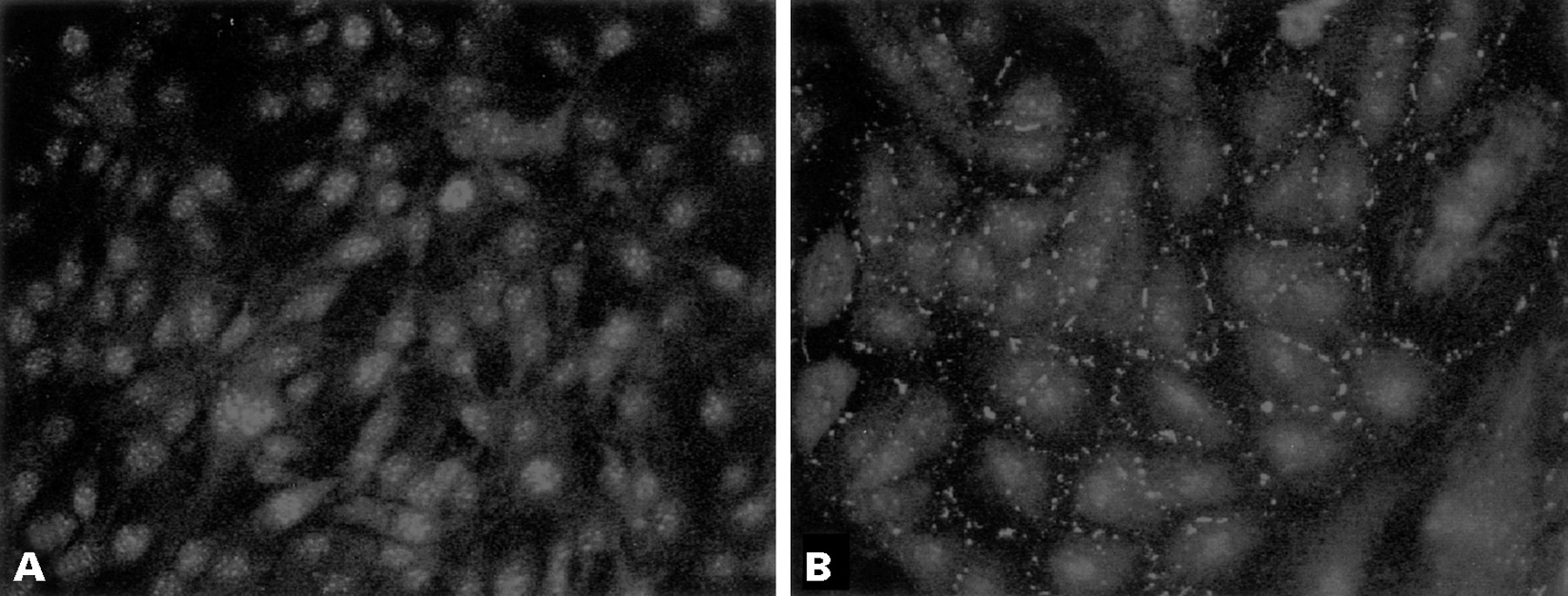
To determine whether the expression of growth control genes could be altered by gap junctional coupling, we pursued a differential display strategy (fig 1). Changes in gene expression between control rat C6 glioma cells and C6 cells transfected with Cx43 were previously detected.25 Using this approach, we determined that some members of the CCN gene family were upregulated. We specifically examined rCCN3 (NOV) expression in C6 glioma cells and transfectants. Whereas rCCN3 (NOV) mRNA species could not be detected by northern blot analysis in C6 glioma cells, they were readily detected in the Cx-13 cell clone, which expresses high amounts of Cx43 and GJIC (fig 2). Immunocytochemical analysis revealed rCCN3 (NOV) to be localised in the cytoplasm and at areas of cell–cell contact in the Cx-13 cells. In contrast, non-transfected parental C6 cells displayed only nuclear staining (fig 3).

Differential display strategy to identify genes altered in C6 glioma cells transfected with connexin 43 (Cx43). Results for four different sets of primers (1, 2, 3, and 4) are shown, with duplicate lanes in each case for C6 cells (6) and Cx-l3 cells (13). Bands were excised from the differential display gel and used as probes on a northern blot containing RNA from C6 cells (C6, C6LTR) and various clones of C6 cells transfected with Cx43 (Cxl2, Cxl4, Cxl3). The degree of Cx43 expression correlates with the level of dye coupling. Arrowheads indicate candidates that are induced by Cx43 expression, whereas arrows show candidates whose expression is reduced in Cx43 expressing cells; modified from Zhu et al.22

Northern blot analysis of CCN3 (NOV) expression in C6 cells and C6 clones expressing connexin 43 (Cx43). CCN3 (NOV) is only expressed in the Cx-13 clone, which shows the highest expression of Cx43 and the greatest degree of gap junctional coupling.
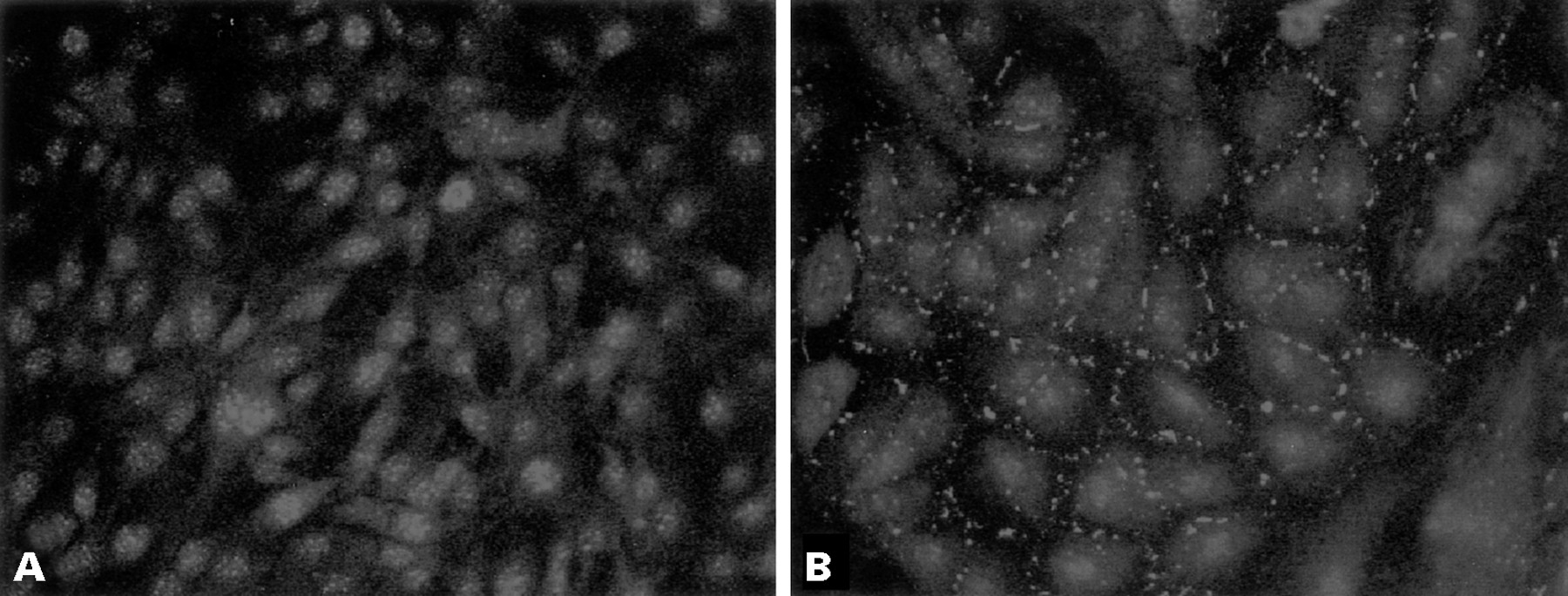
Immunocytochemical analysis of CCN3 (NOV) in (A) C6 cells and (B) Cx-13 cells. C6 cells show nuclear staining, whereas Cx-13 cells show cytoplasmic staining and localisation of CCN3 (NOV) at areas of cell contact. Original magnification, ×250.
EXPRESSION OF CCN3 (NOV) IN HUMAN GLIOMA CELLS MODULATES THEIR TUMORIGENIC POTENTIAL
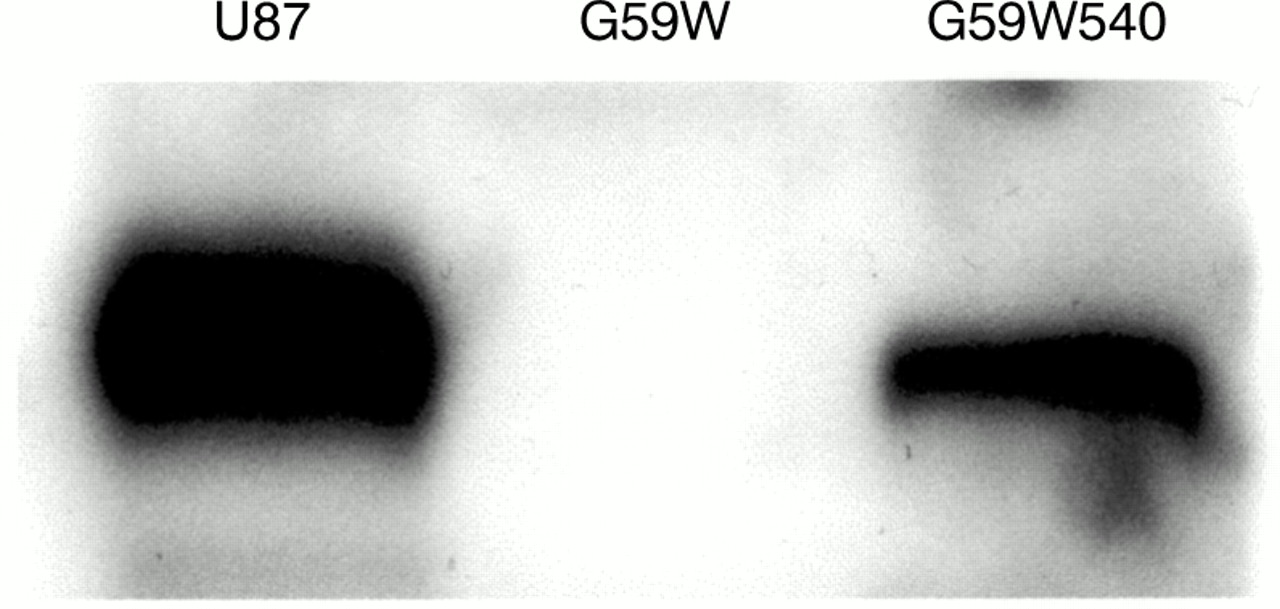
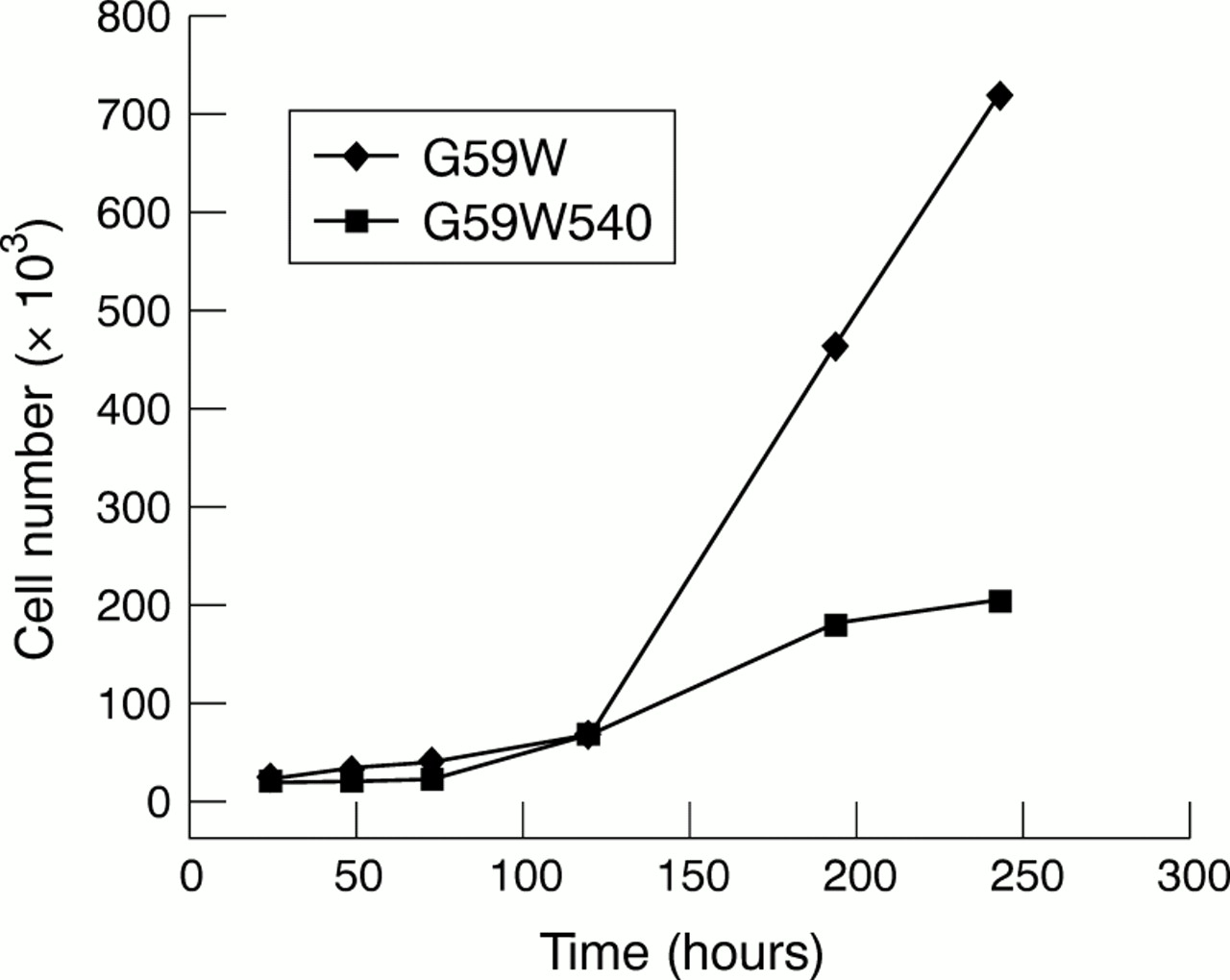
We had established previously that G59W human glioma cells do not express detectable amounts of hCCN3 (NOV).19 Because the results obtained in C6 glioma cells (see above) indicated that the expression of CCN3 (NOV) was upregulated under increased gap junctional intercellular communication, we compared the growth properties and tumorigenic potential of G59W and G59W540 cells. Concentrations of secreted hCCN3 (NOV) were confirmed by immunoblotting of conditioned medium obtained from cells (fig 4). Cells expressing hCCN3 (NOV) (G59W540) showed reduced growth when compared with cells not expressing hCCN3 (NOV) (fig 5). This difference occurred during the exponential phase of growth. The calculated doubling time was 18 hours for the parent cell line and 23 hours for the hCCN3 (NOV) expressing cell line (G59W540). Cell lines expressing intermediate amounts of hCCN3 (NOV) showed corresponding intermediate growth levels (data not shown). We studied whether differences in cell cycle distribution would reflect this change in growth but no significant difference in cell cycle ratios was observed (not shown).

Immunoblot of conditioned medium from glioma cells. Samples (40 μl) of heparin-sepharose concentrated conditioned medium from U87, G59W, and G59W540 glioma cells were electrophoresed and blotted. As previously reported,19 G59W cells do not express detectable amounts of CCN3 (NOV), whereas U87 and G59W540 cells (stably transfected with the pCMV82nov vector) express the full length CCN3 (NOV) protein.
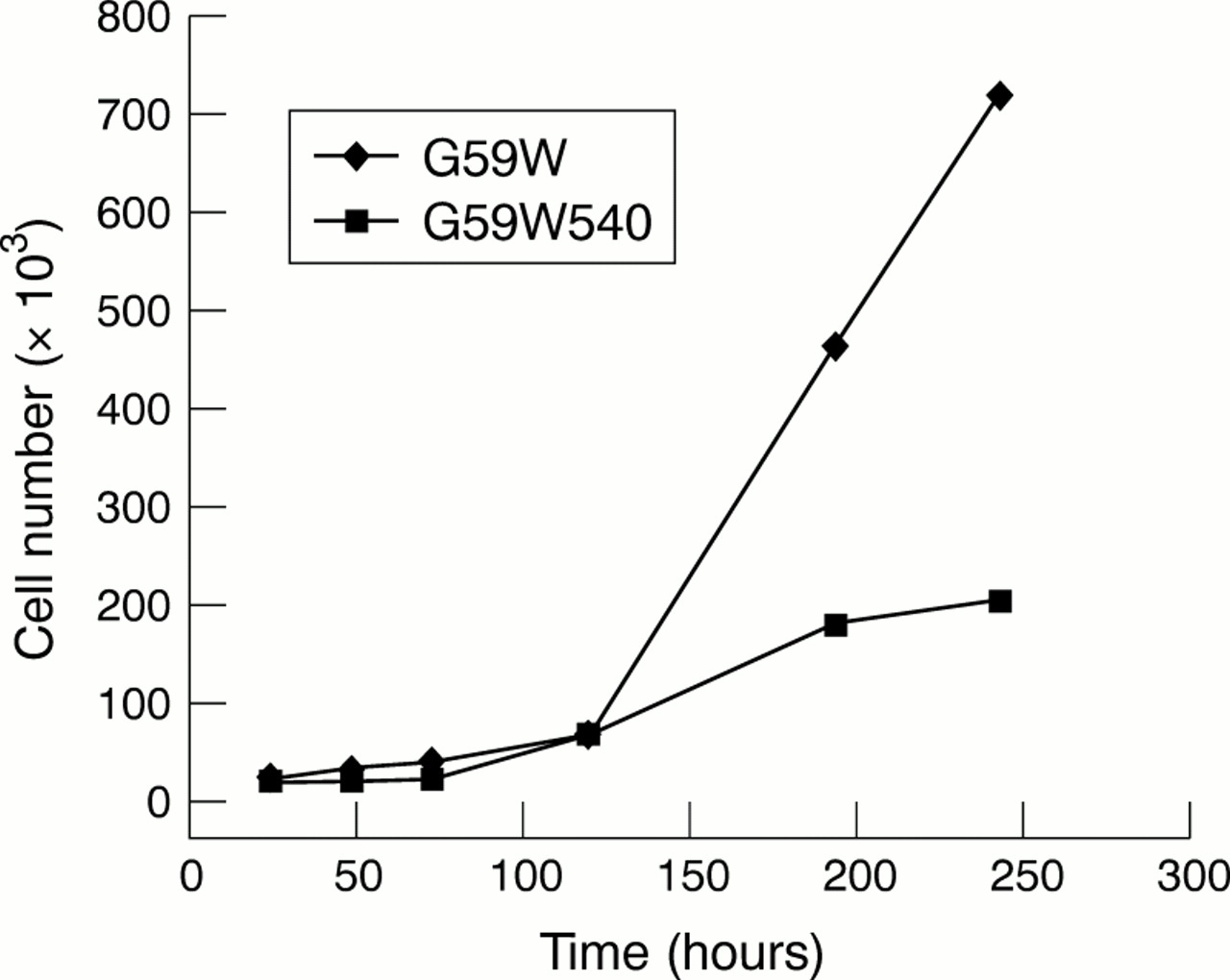
In vitro growth curves of G59W and G59W540 glioma cells. For the in vitro proliferation assay, 20 000 cells from both the G59W and G59W540 cell lines were plated in a six well plate. For each time point, three samples were available for each cell line. Cells were counted at 18 hours, 24 hours, 48 hours, 72 hours, five days, seven days, and nine days.
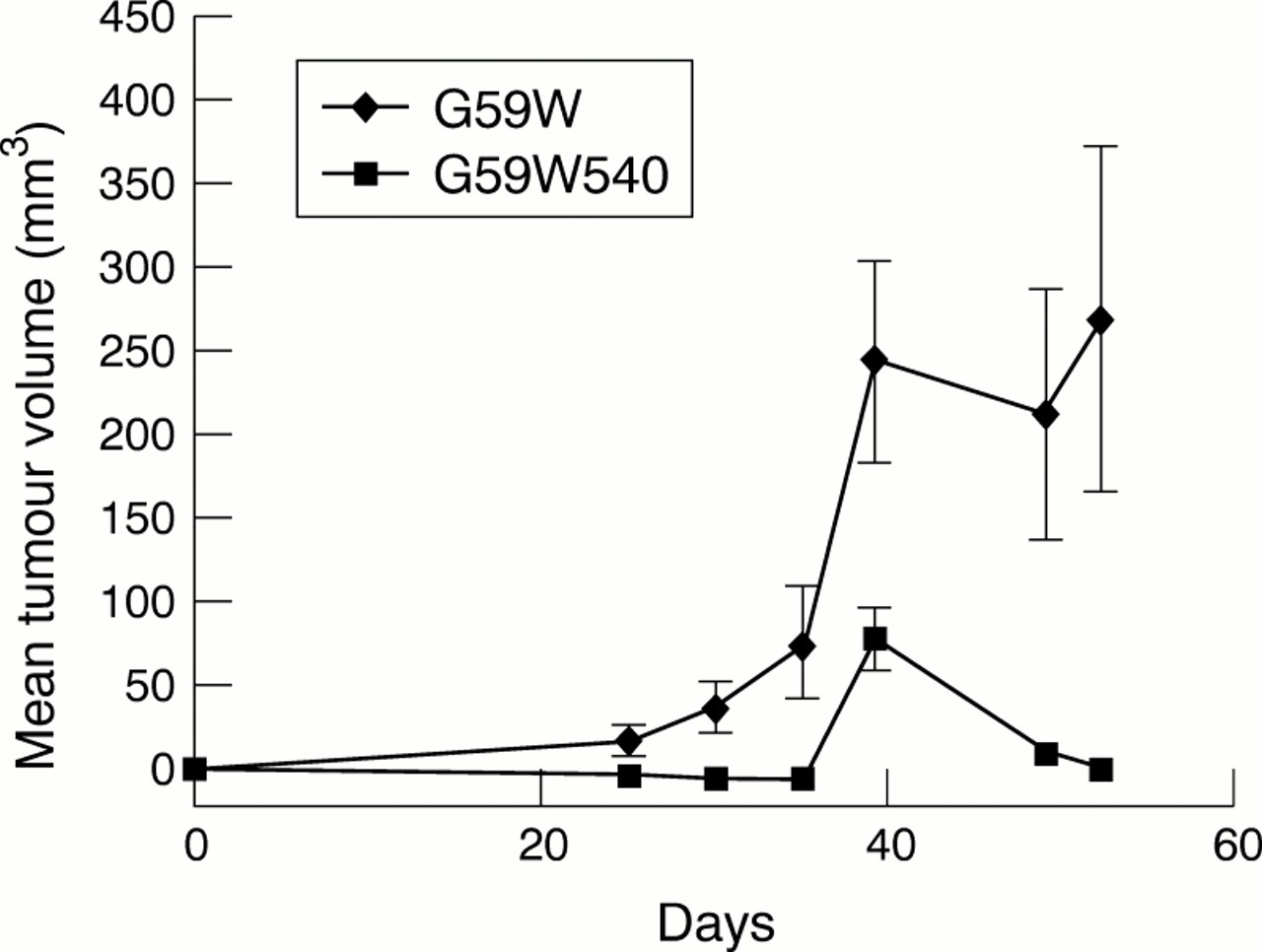
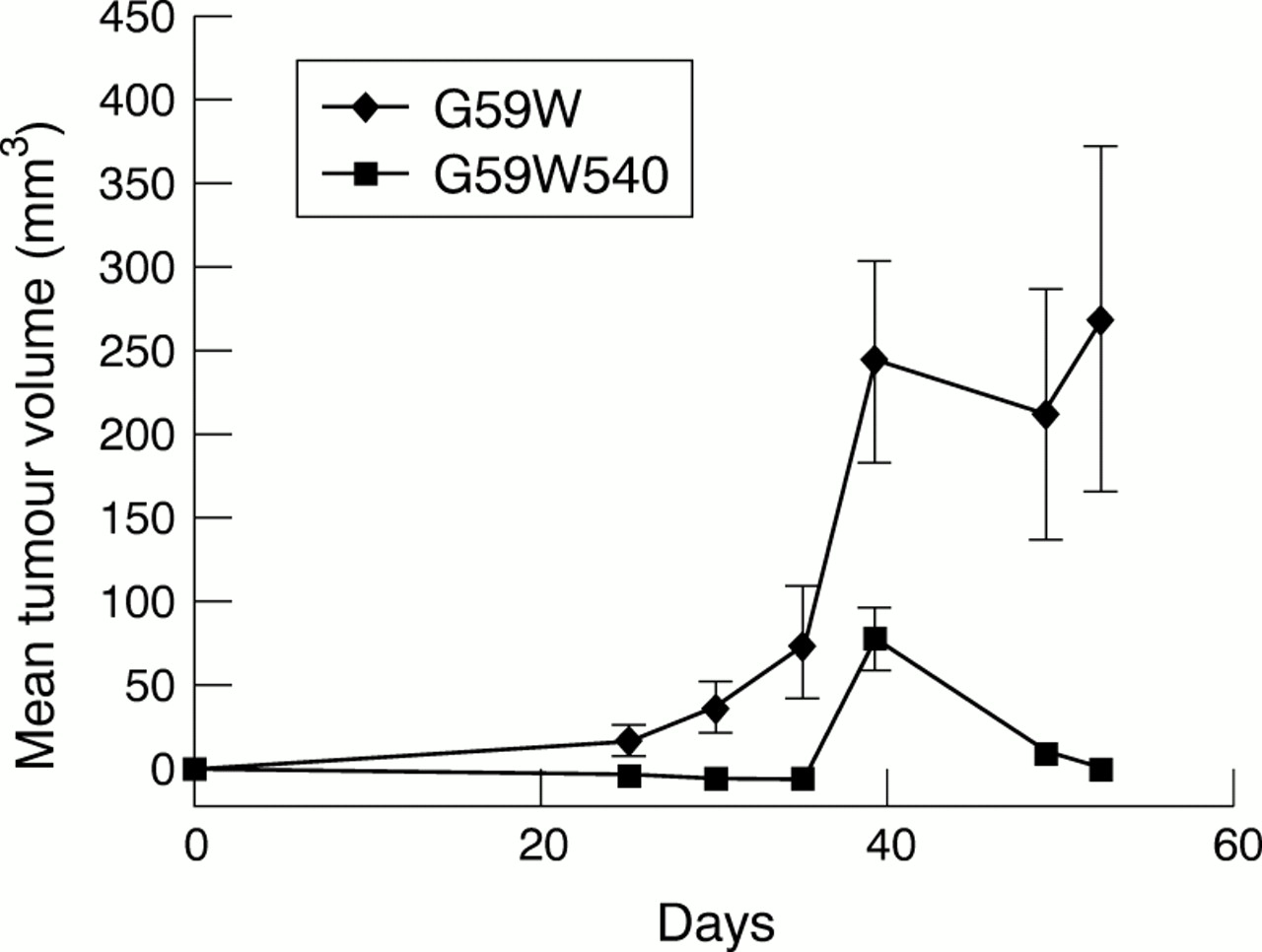
hCCN3 (NOV) overexpression in vitro appeared to have a modest effect upon cell growth, therefore we decided to investigate whether its expression affected the growth of tumours in a xenograft model. The growth of tumours in animals requires a host of additional events such as angiogenesis, invasion of host tissues, and interactions with normal surrounding cells. In nude mice bearing G59W tumours, the tumours reached a volume of 250 mm3 after 40 days (fig 6). In comparison, the animals bearing G59W540 tumours (producing hCCN3 (NOV)) failed to demonstrate any visible growth until at least day 20. Thereafter, tumour growth remained at a plateau, less than 100 mm3, until the end of the study (day 50). The maximal time of tumour growth occurred in G59W cells 30 days after tumour implantation, suggesting that hCCN3 (NOV) inhibits the later development of tumours beyond a threshold size. In addition, 70–80% of mice injected with cells expressing low amounts of hCCN3 (NOV) developed tumours, whereas only 30% of mice injected with cells expressing high amounts of hCCN3 (NOV) developed tumours. Immunohistological examination of the excised tumours and measurement of vascular density by CD31 positive cell counts did not demonstrate an increase in vascularity in the hCCN3 (NOV) expressing tumours (not shown).
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
In vivo growth curves of tumours induced by G59W and G59W540 glioma cells. Two complete animal experiments were performed with G59W and G59W540 cells; 5 × 106 cells were injected into the right hind limb of each mouse with 1/1 medium and metri-gel. Tumour growth was assessed by twice weekly measurements with a caliper; 10 animals were injected in each group. For the G59W cells, all 10 developed tumours. For the G59W540 cells, only three of the 10 animals developed tumours. In each group, the mean size was determined by averaging measurements from the animals that grew tumours. For the second in vivo experiment (data not shown), eight of the 10 animals injected with G59W cells grew tumours, but four of these regressed by the final measurement date (day 53). Of the 10 animals injected with G59W540 cells, only four grew tumours, which were all small.
Discussion
Deciphering the molecular basis for the uncontrolled proliferation of cancer cells has been and remains a very promising avenue in oncology research for the development of new treatments.
Normal cell growth and development require a complex network of communication with surrounding cells, the control of which is dependent upon many different types of regulatory ligands and proteins. The discovery of the CCN family of secreted cell growth regulators has shed new light on some aspects of cell growth regulation. The CCN1 (CYR61) and CCN2 (CTGF) proteins are positive effectors of epidermal growth factor and transforming growth factor β, and recent observations indicate that their activity is mediated through interactions with integrins αvβ3, αIIbβ3, and α6β1.26–28 CCN3 (NOV) was the first example of a CCN member exhibiting a negative effect on cell growth when ectopically expressed as a full length protein. However, the overexpression of full length CCN3 (NOV) in avian and human tumours is frequently seen,1,6 and the addition of recombinant CCN3 (NOV) protein to NIH3T3 cells was reported to stimulate their growth.16 It therefore appears that the functions of the CCN3 (NOV) proteins might be dependent upon the cellular context in which they are expressed. Similar conclusions could be drawn from results obtained with the CCN4 (ELM1/WISP1) and CCN5 (rCOP1/WISP2/CTGFL) proteins,6 the biological activities of which were found to be cell type dependent. Based on these observations, it was proposed that the spatiotemporal control of CCN protein function might be dependent upon distinct combinatorial events, based on the bioavailability of different interacting partners.6
To determine whether the CCN3 (NOV) protein might be useful as a tool in molecular medicine, we have initiated studies aimed at establishing more precisely the biological properties of this protein in tumour cells.
The choice of glioblastomas as a model for studying the antiproliferative potential of CCN3 (NOV) was based upon the initial observation that in glioma and astrocytoma cell lines established from a panel of several human tumours, hCCN3 (NOV) expression was inversely correlated with the tumorigenic potential of the cells.19 This observation, and the previous results that we obtained suggesting that CCN3 (NOV) may act as a negative regulator of cell growth,1,15 led us to investigate more thoroughly the relation between CCN3 (NOV) and tumorigenicity.
Our present results establish that in addition to a negative activity on murine and human cell growth, CCN3 (NOV) also has an antiproliferative effect on tumour cells in vivo.
Although many factors contribute to the transformed phenotype, it has been proposed that the increased growth rate of tumour cells is a direct consequence of the reduction of functional gap junctions in these cells. Indeed, the relation between GJIC and growth rate is well documented,24 and it appears that loss of GJIC might be crucial in the acquisition of metastatic potential. Our results indicate that there is a relation between the establishment of functional GJIC and the expression of rCCN3 (NOV), and suggest that the antiproliferative activity of this protein might involve reorganisation of cellular contacts. Immunofluorescence experiments performed with MDCK cells transfected with pCMV82nov revealed that the hCCN3 (NOV) expressed by these cells localised as patches at the surface (G Chevalier et al, 1997, unpublished). Experiments in progress should establish whether there is a direct interdependence between the formation of gap junctions and the expression of CCN3 (NOV).
The mechanism by which CCN3 (NOV) exercises its negative effect upon the growth of tumour cells in vivo remains to be established at the molecular level. Our present results suggest that CCN3 (NOV) interferes with maintenance, rather than establishment, of the tumour state. The late stages in tumour development require increased angiogenesis. Earlier studies indicated that two members of the CCN family, namely murine CCN1 (CYR61) and CCN2 (FISP12), were potent angiogenesis inducers.29,30
We have reported previously that hCCN3 (NOV) also interacts with fibulin 1C,14 an extracellular protein believed to play a key role in the regulation of cell attachment. The inverse relation that exists between the concentrations of mRNA encoding these two proteins had already suggested that the expression of hCCN3 (NOV) induces, directly or indirectly, the downregulation of fibulin 1C expression.6,14 Fibulin 1C itself binds to components of the extracellular matrix such as fibronectin, laminin, nidogen, and fibrinogen. Therefore, it could be hypothesised that CCN3 (NOV) affects the interaction of cells with the extracellular matrix through intermediary molecules, such as fibulin 1C. Thus, hCCN3 (NOV) could represent a class of tumour suppressor gene that negatively regulates events (such as migration) that require cell–cell and cell–matrix interactions, and thereby cell proliferation of anchorage dependent cells. We have reported previously that CCN3 (NOV) is a marker of differentiation in several tumour types, such as Wilms's tumours and musculoskeletal tumours,6,14 and a marker of poor prognosis in other tumours, such as renal cell carcinoma31 and Ewing's tumours6 (Manara et al, 2001, unpublished). Our demonstration that the secreted form of CCN3 (NOV) may suppress tumour growth opens up the possibility of CCN3 (NOV) as a potentially effective anticancer agent.
Acknowledgments
We thank Professor M Westphal for providing the G59W cells, Dr P Ellis for providing the antirat CCN3 (NOV) antibody, and Dr H Yeger for critical reading of the manuscript. The authors wish to acknowledge Dr B Roizman for support and helpful discussions. M Laurent is acknowledged for technical assistance with cell cultures. These studies were supported in part by grants from the ARC and the Ligue Nationale Contre le Cancer (Comités du Cher et de l'Indre) to B Perbal, and the Canadian Institutes of Health Research (MOP-14358) to CC Naus.