Article Text

Abstract
INTRODUCTION Hypermethylation of the promoter region of the hMLH1 gene is associated with absent expression of MLH1 protein in sporadic colorectal cancers with microsatellite instability (MSI+), and it has been proposed that methylation may be a mechanism of inactivation in Knudson's hypothesis. The incidence of hypermethylation of thehMLH1 promoter in hereditary non-polyposis colorectal cancer (HNPCC) versus MSI+ sporadic colorectal cancer was investigated and compared.
METHODS DNA was available from 10 HNPCC colorectal cancers (median age 58 years, range 39-67) with germline mutations in hMLH1 and 10 MSI+ sporadic colorectal cancers (mean age 79 years, range 41-85). MSI was determined by amplification of BAT26 and TGF-β RII. The methylation status of the hMLH1 promoter was studied by the polymerase chain reaction (PCR) basedHpaII restriction enzyme assay technique. Evidence of allelic loss at hMLH1 was searched for in the HNPCC colorectal cancers.
RESULTS All cases were confirmed to be MSI+. The promoter region of hMLH1 was hypermethylated in seven of 10 MSI+ sporadic cancers versus 0 of 10 HNPCC cancers (p<0.002). Evidence of loss of heterozygosity athMLH1 was observed in eight of the 10 HNPCC colorectal cancers.
CONCLUSION While mutations and allelic loss are responsible for the MSI+ phenotype in HNPCC cancers, the majority of MSI+ sporadic cancers are hypermethylated in the promoter region of hMLH1. These data further support our argument that tumours from HNPCC patients, which almost always acquire a raised mutation rate, mostly follow a different pathway from MSI+ sporadic tumours.
- hMLH1 promoter region
- HNPCC
- hypermethylation
- colorectal cancer
Statistics from Altmetric.com
Hereditary non-polyposis colorectal cancer (HNPCC) is caused by germline mutations in the DNA mismatch repair (MMR) genes.1 To date, inactivating mutations have been described in five mismatch repair genes: hMSH2, hMLH1, hPMS1, hPMS2, and GTBP(hMSH6).2-9 These cancers, from affected HNPCC kindreds, exhibit genomic instability which can be detected as changes in the length of microsatellite sequences.10 Almost all colorectal cancers (CRC) removed from patients with HNPCC show microsatellite instability (MSI+) or are replication error positive (RER+),10 and this contrasts with approximately 15-20% of sporadic CRC from patients with no obvious family history.11 Approximately 70% of HNPCC patients with MSI+ tumours are found to have germline mutations in one of the mismatch repair genes.12 The majority of these mutations (95%) have been described inhMLH1 andhMSH2,13 with mutations in other mismatch repair genes being rare.13 In contrast, somatic mutations in the mismatch repair genes are rarely described in MSI+ sporadic CRC.14 15 Epigenetic mechanisms which inactivate genes, such as promoter region hypermethylation resulting in transcriptional loss, have been described in a number of tumour suppressor genes,16 and recently it has been shown that hypermethylation of the promoter region of thehMLH1 gene may cause a lack of expression of its protein and therefore account for microsatellite instability in MSI+ sporadic CRC.12 17-20
With the recognition that methylation of the promoter region may inactivate tumour suppressor genes, Jones and Laird21 have proposed that Knudson's hypothesis, for the inactivation of tumour suppressor genes, should be expanded to include epigenetic mechanisms of gene inactivation, such as hypermethylation of the promoter region. We have therefore studied a panel of HNPCC CRC, and compared this to a panel of MSI+ sporadic CRC, to determine whether hypermethylation of the hMLH1 promoter region is a common “second hit” mechanism of inactivation in both of these two cancers.
Methods
MATERIALS
Tumour DNA was available from 10 HNPCC family members with CRC (median age 58 years, range 39-67) and from 10 MSI+ sporadic CRC (mean age 79 years, range 41-85), at the University of Helsinki, Finland. The MSI status of all tumours was determined by amplification of BAT26 and TGF-β RII mononucleotide markers as previously described.22 The HNPCC family members all carried germline mutations in hMLH1. This included six patients with the Finnish founder mutation 1 (a 3.5 kb genomic deletion of hMLH1 comprising exon 16) detected by PCR specific for the normal and mutated allele which are then size fractionated by agarose gel electrophoresis.23 Three patients had the Finnish founder mutation 2 (a single bp substitution, cagGTG→caaGTG, at the exon 6 acceptor splice site inhMLH1), which was detected by allele specific oligonucleotide hybridisation,23 and one patient had a nonsense mutation in exon 4 of hMLH1detected by direct sequencing (codon 126, TAC→TAG, Tyr→Stop). No mutations were detected in the germline DNA from patients with MSI+ sporadic cancers, which were directly sequenced throughout the coding region (including the intron/exon borders) and the promoter region of hMLH1and hMSH2.
METHYLATION STATUS
Hypermethylation of the hMLH1 promoter region was ascertained by a PCR based HpaII restriction enzyme assay.12 A total of 250 ng of genomic DNA from each tumour was digested separately byHpaII and MspI in 20 μl volumes of restriction enzyme buffer (New England Biolabs). DNA from each sample was also added to 20 μl of restriction enzyme buffer without enzyme, as a control. A total of 25 ng of DNA from each digest was analysed by PCR in 50 μl reactions using conditions and primers, for the promoter region of hMLH1, as previously described.12 PCR products were observed by agarose gel electrophoresis. Fisher's exact value was calculated for the incidence of hypermethylation of the hMLH1promoter region in HNPCC cancers versus MSI+ sporadic cancers.
LOSS OF HETEROZYGOSITY (LOH) ANALYSIS
LOH studies were performed on all 10 HNPCC cancers. Owing to slippage at DNA repeat sequences in MSI+ cancers and subsequent difficulty in the interpretation of results, we did not use CA repeat markers to study LOH. For mutation 1, radiolabelled PCR using previously described primers, specific for the normal and mutated allele,23 was performed on tumour and normal DNA and the alleles were size fractionated by polyacrylamide gel electrophoresis. The intensities of each band were analysed, using the BAS-MP2040 Fujifilm Imaging Plate for Bio Imaging Analyzer (Fuji Photo Film Co Ltd, Japan) in six positions (two positions centrally and four positions from the perimeter) and a mean value calculated. The ratio of the normal (T1) and mutant (T2) bands from the tumour (T1:T2, where T1 is the smaller value) was compared to the germline ratio (N1:N2, where N1 is the smaller value) and an allele ratio obtained (T1:T2/N1:N2). LOH was declared when the allele ratio was less than 0.6 or greater than 1.67, as previously defined.24
For mutation 2 and the exon 4 nonsense mutation, quantification of allelic loss was more difficult, but evidence of LOH was declared in the presence of a higher wild type:mutant allele ratio in normal DNA when compared to tumour DNA following direct sequencing.
Results
The clinicopathological features of the HNPCC cancers and MSI+ sporadic cancers are shown in table 1. The HNPCC cancers, as expected, had a younger median age, 58 years versus 79 years, at presentation (Mann Whitney U test, 0.01<p<0.05). All cancers were confirmed as MSI+.
HNPCC versus MSI+ sporadic CRC
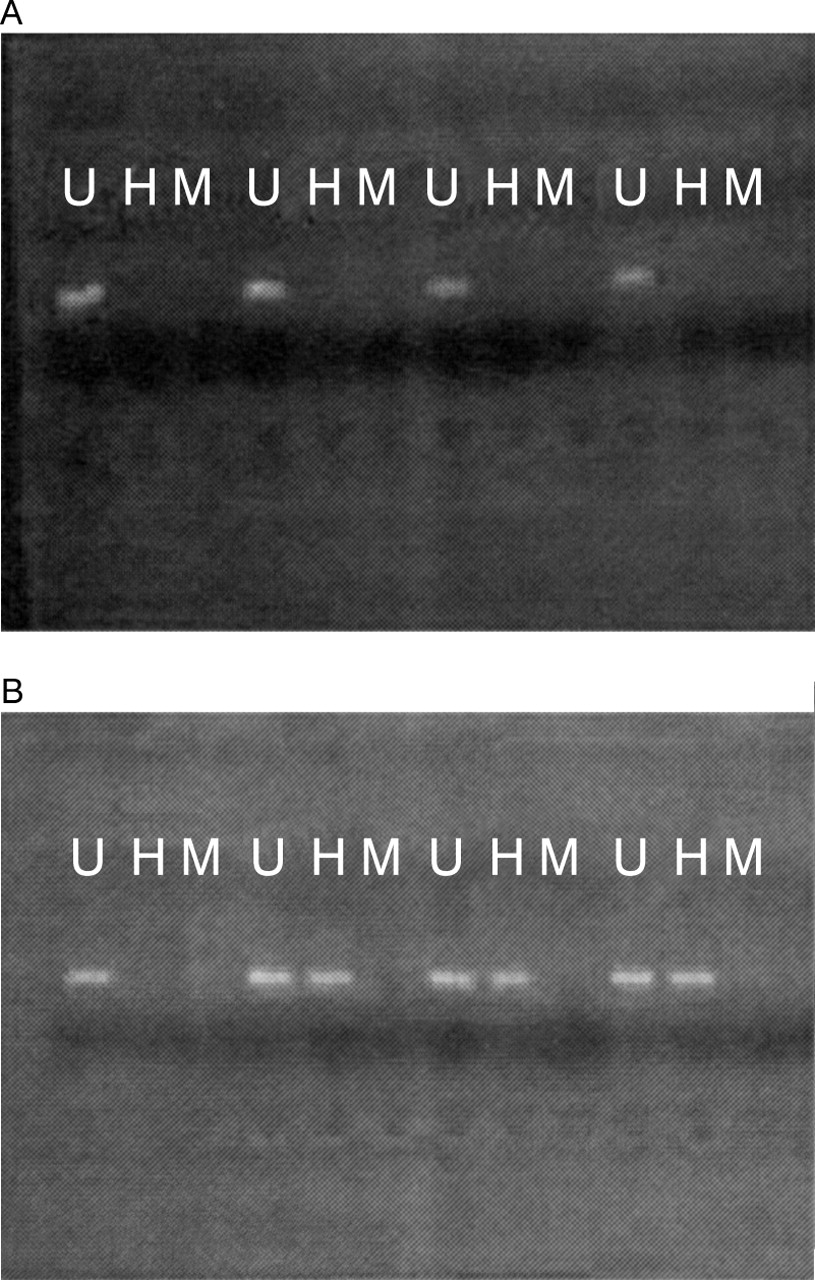
The promoter region of hMLH1 was hypermethylated in seven of 10 MSI+ sporadic cases versus 0 of 10 HNPCC cases (Fisher' exact test, p<0.002) (fig 1).

Representative results from PCR based HpaII restriction enzyme assay. All 10 HNPPC cancers (A) were unmethylated in the hMLH1 promoter region, while seven of 10 MSI+ sporadic cancers (B) were methylated (p<0.002). A PCR product is seen following incubation with HpaII in a methylated cancer, as the methylated restriction site (CCGG) is resistant to digestion by the enzyme (U, undigested; H, incubated with HpaII; M, incubated with MspI).
There was at least some evidence of loss of heterozygosity athMLH1 in eight of the 10 HNPCC colorectal cancers (table 2). Radiolabelled PCR showed evidence of LOH in four of five cancers from patients with the Finnish founder mutation 1 (the remaining cancer was uninformative). The other four patients showed evidence of LOH on direct sequencing (fig2).
Mean allele ratio for Finnish founder mutation 1 using radiolabelled PCR specific for the normal and mutated allele, which are then size fractionated by agarose gel electrophoresis and analysed for intensities of mutant and wild type alleles

{kind=link}
{kind=link}
For Finnish founder mutation 2, loss of heterozygosity (LOH) was declared in the presence of a higher wild type:mutant allele ratio in normal DNA when compared to tumour DNA following direct sequencing. Reverse sequencing of hMLH1, exon 6, from a patient with the founder mutation 2 (a single bp substitution, cagGTG→caaGTG, at the exon 6 acceptor splice site in hMLH1) is shown. A higher wild type:mutant allele ratio (arrows) is seen in the normal DNA (above) when compared to the tumour DNA (below).
Discussion
None of the 10 HNPCC cancers were hypermethylated in thehMLH1 promoter region, and these results suggest that methylation is not a common “second hit” mechanism in the inactivation of mismatch repair genes in HNPCC cancers. Although Jones and Laird21 have recently pointed out that Knudson's two hit hypothesis should extend to include epigenetic mechanisms of gene inactivation, such as methylation, this would only appear to be true of MSI+ sporadic cancers in the case ofhMLH1. Hypermethylation of thehMLH1 promoter is responsible for the MSI+ phenotype in as many as 80% of MSI+ sporadic colorectal cancers.17 Hypermethylation of the promoter region has not been reported in hMSH2, although this is not surprising as Thibodeau et al 14 have reported thathMLH1 is the altered protein in more than 90% of MSI+ sporadic CRC. As we had not detected methylation in the HNPCC cancers, we sought evidence of allelic loss as the “second hit”.
As there are technical difficulties with studying allelic loss in MSI+ cancers, we used the germline mutations as specific LOH markers, and were able to detect at least some evidence of LOH at hMLH1 in eight of the 10 HNPCC cancers, and the finding of frequent allelic loss at this locus is in keeping with others.25 26 In contrast, it has been shown that hypermethylation of thehMLH1 promoter region is a biallelic event in MSI+ sporadic CRC and is not accompanied by LOH.18 20
Mutations which are relatively common, such as the Finnish founder mutations, are likely to have arisen many tens or hundreds of generations ago. The pedigrees with founder mutations in this series have been extended two to four generations back (and horizontally) and are known to be unrelated. Although it is conceivable that the selective advantage associated with the two most frequent HNPCC mutations in Finland (nine of 10 cases in this series) leads to a particular second event (LOH and not methylation), this would seem unlikely. Common lineage would not be expected five or 10 generations ago, let alone the time that has passed since these mutations arose.
The underlying event which leads to methylation may be a change in some aspect of chromatin structure possibly connected with histone acetylation,27 which enables recently described methyl transferases28 to have access to the DNA. Previous work has shown that methylation may accompany missense mutations inhMLH1 in MSI+ sporadic CRC,20but it is not clear which event occurs first. Presumably, a missense change precedes methylation or one allele is methylated before the other, otherwise there would be no basis for selection for a missense mutation.
Others have shown that MSI+ sporadic CRC may be a subgroup of tumours which have increased promoter region hypermethylation in a number of genes, including p16 and IGFII.29 Thus, the inactivation of hMLH1 by promoter region hypermethylation may, in turn, enhance the rate of further genetic events in MSI+ sporadic cancers.21
The majority of MSI+ sporadic cancers are hypermethylated in the promoter region of hMLH1, while mutations and allelic loss are responsible for the MSI+ phenotype in HNPCC cancers. These data further support our argument that tumours from HNPCC patients, which almost always acquire a raised mutation rate, mostly follow a different pathway from MSI+ sporadic tumours.30 31 In the latter, we believe that a single mutation or methylated allele initially will be selected for, and that the subsequent event leading to the MSI+ phenotype does not occur particularly early in the adenoma to carcinoma sequence. At this late stage, the mutation rate is less limiting because of increased population size and possibly other constraints on tumour growth. This prediction is supported by experimental evidence.32
Germline mutations in HNPCC patients occur throughouthMLH1 and hMSH2,although nine of 10 mutations in this series arehMLH1 founding mutations originating in Finland.23 33 In contrast, hMLH1is inactivated, whether by mutation or methylation, in the majority of MSI+ sporadic CRC.20 34 This situation is analogous to APC, with the majority of somatic mutations occurring in the mutation cluster region. It is likely that the selective advantage needed to promote tumour outgrowth is much less for germline mutations, which occur in all cells, than for somatic mutations.
Selection for inactivation of a mismatch repair gene may be for resistance to apoptosis, as is the case forp53 mutations,35 and not an increased mutator phenotype. Recently, it has been shown that apoptosis can be induced by the overexpression ofhMSH2 orhMLH1.36 This produces the possibility that HNPCC and MSI+ sporadic cancers lose the ability to undergo efficient apoptosis.
In conclusion, while hypermethylation of the hMLH1promoter is responsible for the MSI+ phenotype in the majority of MSI+ sporadic CRC, it is not a common “second hit” mechanism of inactivation of mismatch repair genes in HNPCC CRC.