Article Text

Abstract
This review focuses on selected areas that should interest both the scientist and the clinician alike: polymorphisms within the factor VIII and factor IX genes, their linkage, and their ethnic variation; a general assessment of mutations within both genes and a detailed inspection of the molecular pathology of certain mutations to illustrate the diverse cause–effect relations that exist; a summary of current knowledge on molecular aspects of inhibitor production; and an introduction to the new areas of factor VIII and factor IX catabolism. An appendix defining various terms encountered in the molecular genetics of the haemophilias is included, together with an appendix providing accession numbers and locus identification links for accessing gene and sequence information in the international nucleic acid databases.
- haemophilia A
- haemophilia B
- factor VIII
- factor IX
- polymorphism
- mutation
- inhibitor
- catabolism
- ARE, androgen response element
- C/EBP, CCAAT/enhancer binding protein
- EGF, epidermal growth factor
- LD, long distance
- LRP, lipoprotein receptor related protein
- MHC, major histocompatibility complex
- PCR, polymerase chain reaction
- RFLP, restriction fragment length polymorphism
- SNP, single nucleotide polymorphism
- VNTR, variable number tandem repeat sequence
Statistics from Altmetric.com
- ARE, androgen response element
- C/EBP, CCAAT/enhancer binding protein
- EGF, epidermal growth factor
- LD, long distance
- LRP, lipoprotein receptor related protein
- MHC, major histocompatibility complex
- PCR, polymerase chain reaction
- RFLP, restriction fragment length polymorphism
- SNP, single nucleotide polymorphism
- VNTR, variable number tandem repeat sequence
The molecular basis of the hereditary haemophilias has been unravelled to a remarkable degree over the past decade. The early studies, which glimpsed the possibility of mutational heterogeneity, have been verified by the almost breath taking diversity of mutations that have since been characterised in the coagulation factor VIII gene in haemophilia A and the coagulation factor IX gene in haemophilia B. Although mutational heterogeneity abounds within these two genes, natural polymorphic heterogeneity does not. These apposite features have made the collection of informative genetic data for familial investigations a difficult task. However, modern techniques of molecular biology have decreased the enormity of the task of mutation analysis in these two genes and have decreased the workload for the analysis of their polymorphisms. This review focuses on polymorphisms and mutations in the genes for coagulation factor VIII and factor IX, molecular biological aspects of inhibitor production, and the new areas of factor VIII and factor IX catabolism. The molecular pathology of certain mutations is dealt with to exemplify the interesting and revealing cause–effect relations that exist within the haemophilias.
“An almost breath taking diversity of mutations have since been characterised in the coagulation factor VIII gene in haemophilia A and the coagulation factor IX gene in haemophilia B”
BACKGROUND: FACTOR VIII AND FACTOR IX IN COAGULATION
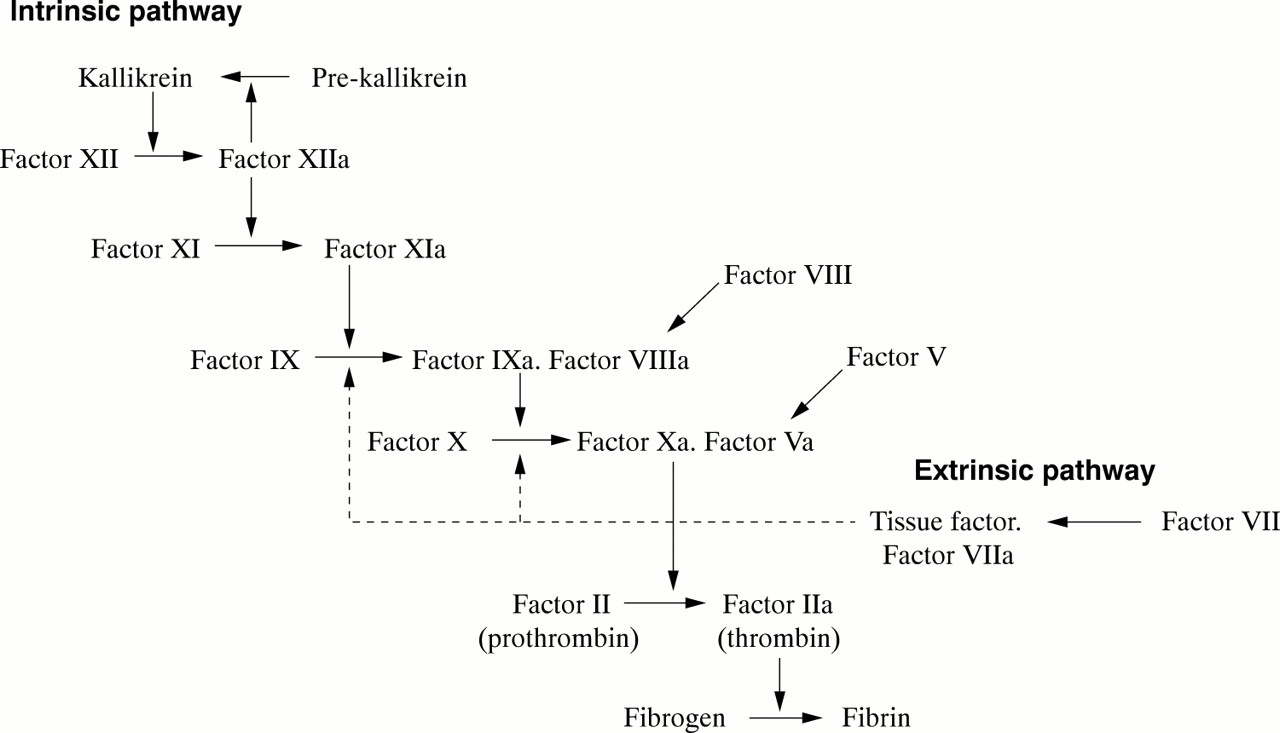
These two proteins circulate as inactive precursors that are activated at the time of haemostatic challenge, via the intrinsic or extrinsic pathways of the coagulation cascade (fig 1). Factor VIII is a protein cofactor with no enzyme activity per se; factor IX is a serine protease with an absolute requirement for factor VIII as cofactor. Upon activation, and in the presence of calcium ions and phospholipid surfaces, factor VIII and factor IX form an active complex, the tenase complex, which activates factor X (fig 1). Subsequent stages of the cascade then proceed, culminating in the deposition of fibrin, the structural polymer of the blood clot.
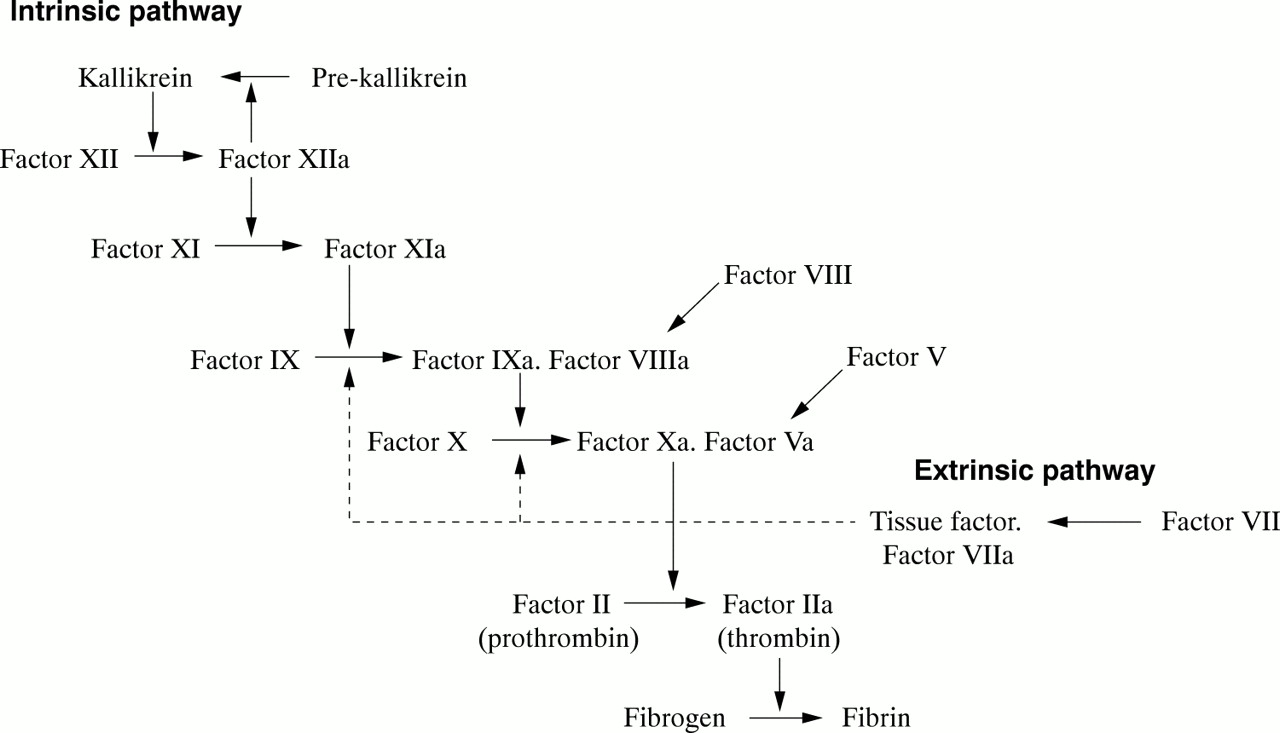
Schematic showing the intrinsic and extrinsic pathways of the coagulation cascade leading to fibrin formation. A deficiency or dysfunction of coagulation factor VIII or factor IX compromises the activation of factor X, the ensuing reactions are inefficient and haemophilia results.
A deficiency or dysfunction of either factor VIII or factor IX compromises the activation of factor X, so that the ensuing steps of the coagulation cascade are also compromised and fibrin deposition is either inefficient or non-existent. Thus, the fundamental biochemical lesion underlying the haemophilias is an insufficiency of the activity of the tenase complex, brought about either by a deficiency of coagulation factor VIII cofactor activity (haemophilia A) or coagulation factor IX enzyme activity (haemophilia B). Thus, it is not surprising that the two disorders are clinically similar because they both arise from perturbation of the same essential step in the process of fibrin generation.
THE FACTOR VIII AND FACTOR IX GENES AND PROTEINS
Both genes are located on the X chromosome: the factor VIII gene is located towards the end of the long arm at Xq281; the factor IX gene is also located on the long arm, more towards the centromere at Xq27.2 There is a considerable genetic distance (0.5 cM) between the two genes3,4; they are by no means in tight linkage with each other. Thus, haemophilia A and B are X linked disorders; they are recessive, carried by females (karyotype 46:XX), and present in males (karyotype 46:XY). Rare cases of female haemophilia are known5,6 and these may arise through the presence of two defective factor VIII or factor IX genes, or through non-random X chromosome inactivation. The latter has often been offered as a primary explanation for haemophilia symptoms in known carriers; however, recent data suggest that there is an absence of correlation between X chromosome inactivation and plasma concentrations of factor VIII in carriers of haemophilia A and B.7
Factor VIII
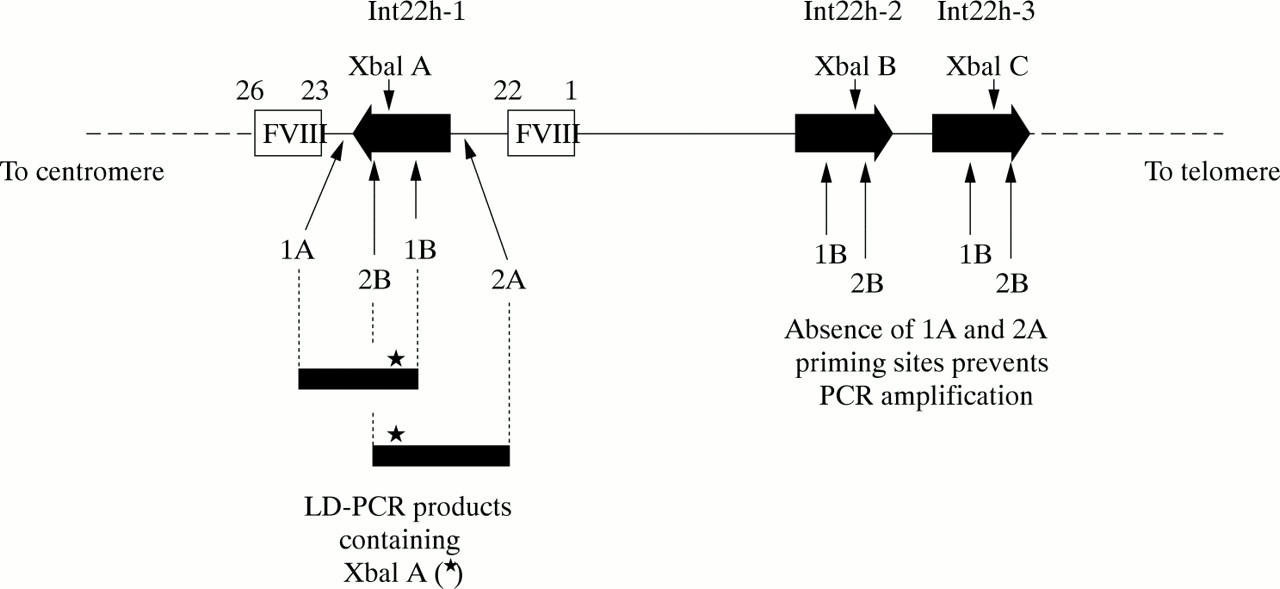
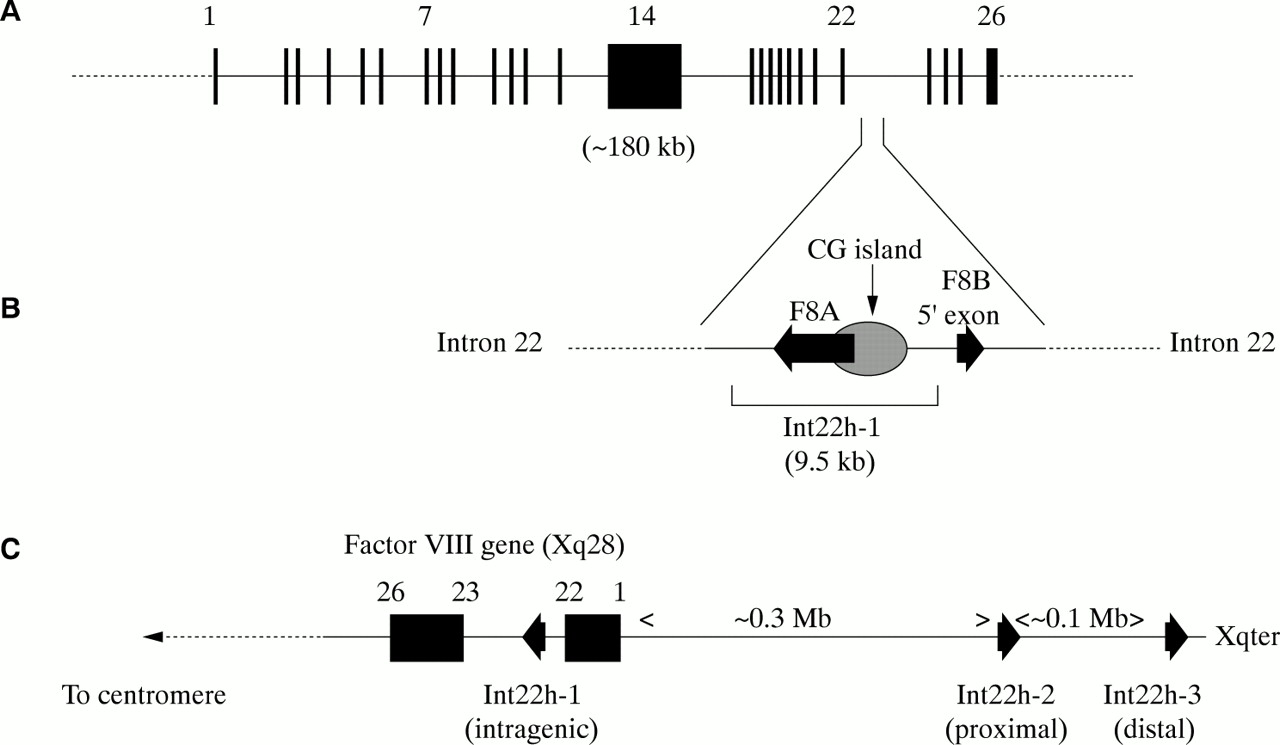
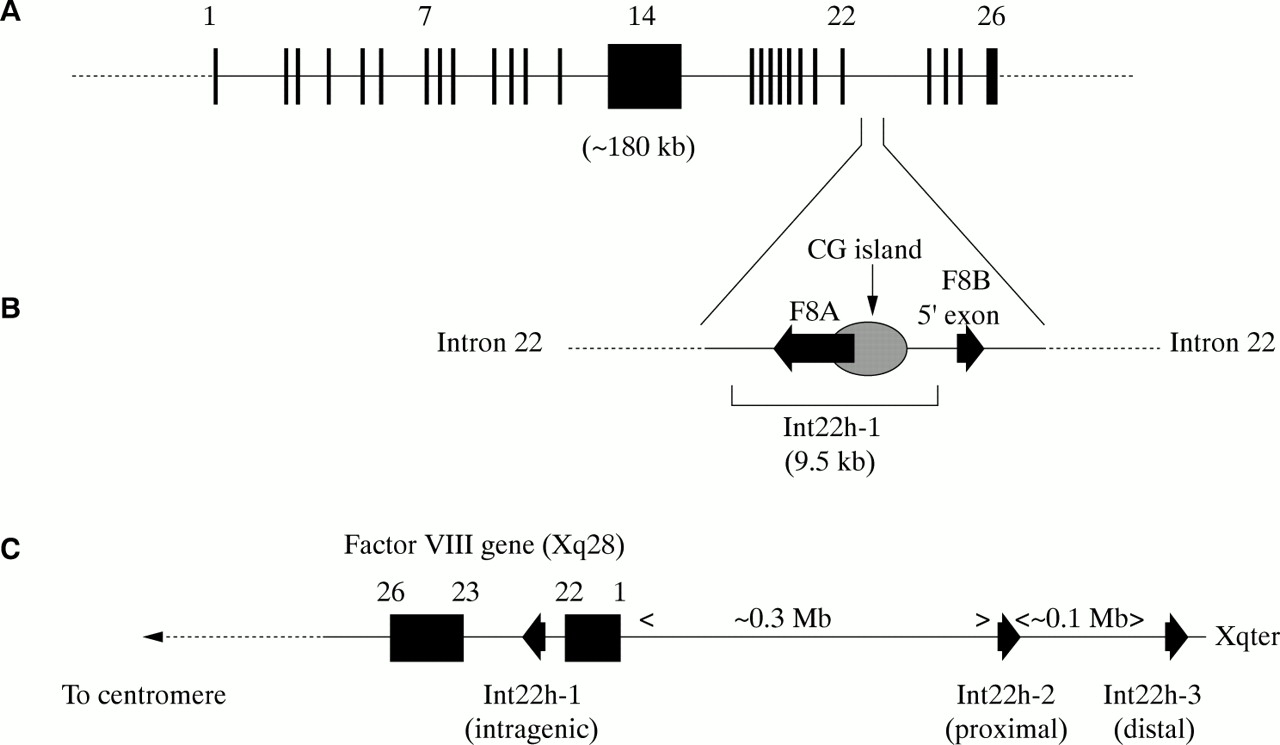
The factor VIII gene is extremely large (∼ 180 kb) and structurally complex (26 exons) (fig 2A; table 1).1 Intron 22 of the gene contains a CpG island, which acts as a bidirectional promoter for two further genes, F8A8 and F8B9 (fig 2B). The CpG island and F8A are contained within a stretch of DNA of approximately 9.5 kb, which is repeated at least twice on the X chromosome, further towards the telomere and extragenic to the factor VIII gene8,10 (fig 2C). These homologues are known as int22h-1 (intragenic) and int22h-2 and int22h-3 (extragenic).10
Exon and intron sizes for the human factor VIII gene1

(A) Genomic organisation of the human factor VIII gene. Exon and intron sizes are given in table 1. (B) Enlargement of a portion of intron 22 of the factor VIII gene to show the relative locations and orientations of the CG island, the intragenic gene F8A, the 5` exon of the putative gene F8B, and the extent of the int22h-1 homologue, which is duplicated towards the telomere of Xq. (C) Orientation of the factor VIII gene within Xq28 and location and orientation of int22h homologues.
F8A is intronless and spans less than 2 kb within intron 22.8 It is transcribed in the direction opposite to factor VIII.8 It is transcribed abundantly in a wide variety of cell types8 and is conserved in the mouse, which implies it has some function.11
F8B comprises a 5` exon located within intron 22 and then exons 23–26 of the factor VIII gene.10 The 5` exon potentially encodes eight amino acids and the factor VIII reading frame is maintained for exons 23–26.10 The result is a protein that is considerably shorter than factor VIII and that includes the phospholipid binding domain.
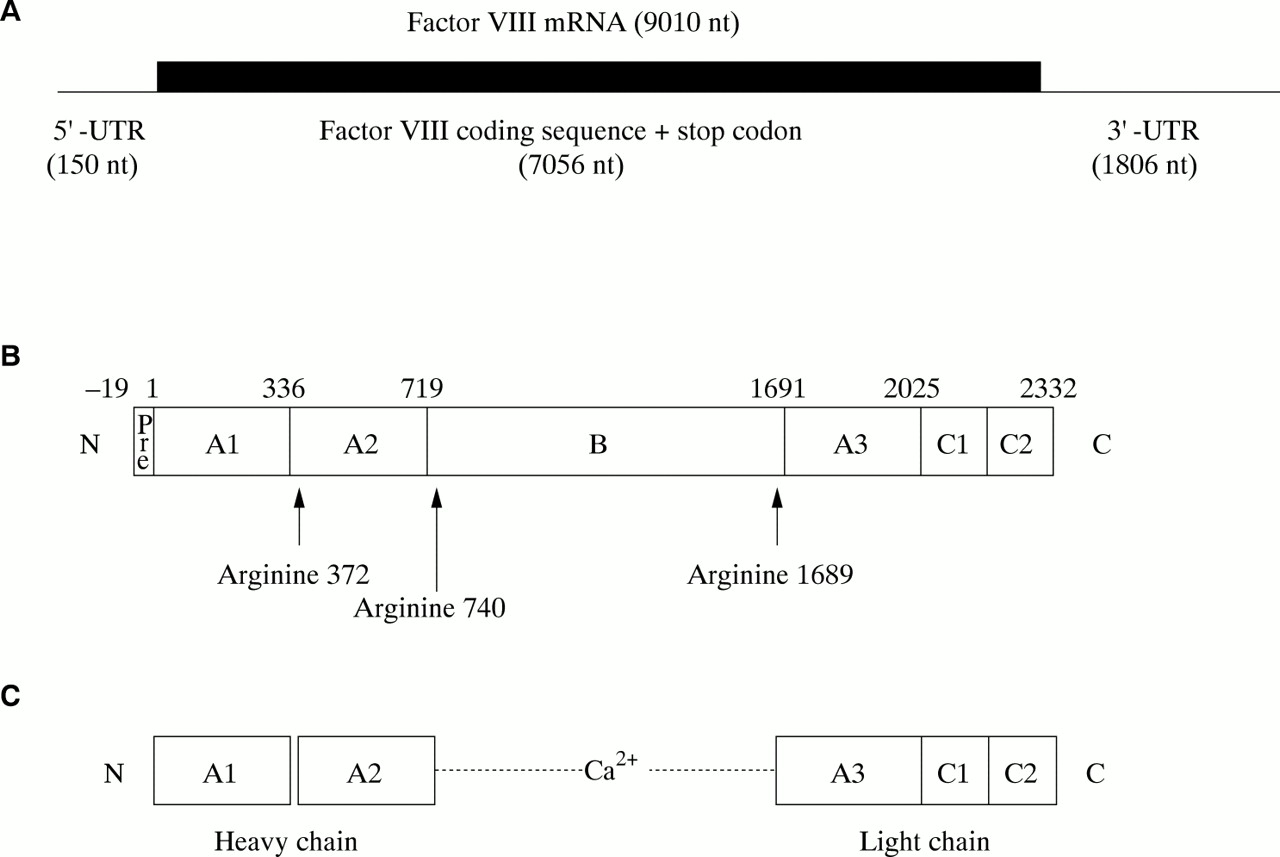
The factor VIII gene transcript is approximately 9010 bases in length and comprises a short 5` untranslated region (150 bases), an open reading frame plus stop codon (7056 bases), and an unusually long 3` untranslated region (1806 bases)12 (fig 3A). The open reading frame encodes a signal peptide of 19 amino acids, which directs the passage of factor VIII through the cell, and a mature protein of 2332 amino acids, which is the circulating inactive pro-cofactor (fig 3B).12 This contains a distinct domain structure based on homologies within the protein.13
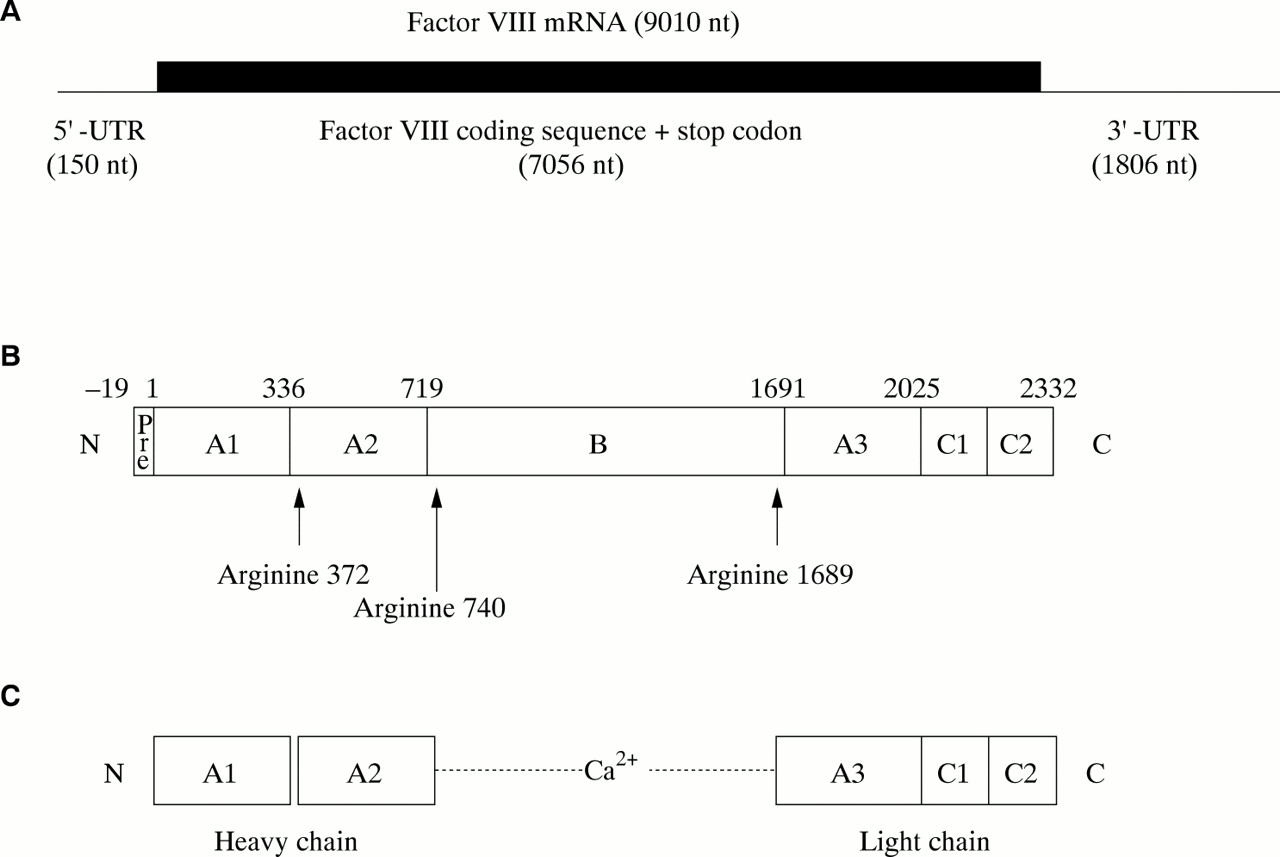
(A) Factor VIII mRNA showing the extent and location of the open reading frame. (B) The newly synthesised factor VIII protein molecule comprising a pre-sequence of 19 amino acids and a mature peptide of 2332 amino acids (total length, 2351 amino acids). A1–3, B, C1, and C2 represent domains assigned according to homologies within the protein (the boundaries are somewhat arbitrary). The arginine residues signalling the sites for proteolytic activation are arrowed. (C) Activated factor VIII comprising a heterotrimer in which the dimeric N-terminal heavy chain is held together with the monomeric C-terminal light chain by a metal ion bridge (Ca2+).
Factor VIII is activated by proteolysis catalysed by thrombin (fig 3C) (reviewed in Tuddenham and Cooper14). The principal activation cleavage sites are on the C-terminal side of arginine residues 372, 740, and 1689.15 Activated factor X also cleaves at these sites. The activation cleavage sites flank the B domain, which is released from factor VIII on activation, leaving a heterotrimer comprising an N-terminal heavy chain and a C-terminal light chain; these are held together by Ca2+. In the circulation, factor VIII is carried and protected by von Willebrand factor.16,17 Proteolytic cleavage of factor VIII at the time of activation simultaneously releases it from its complex with von Willebrand factor.18,19
“In the circulation, factor VIII is carried and protected by von Willebrand factor”
Factor IX
The factor IX gene is considerably smaller and structurally simpler than that for factor VIII. It is approximately 34 kb in length and contains only eight exons, the largest of which is only 1935 bp (fig 4A; table 2).2 The transcript is 2803 bases in length and comprises a short 5` untranslated region (29 bases), an open reading frame plus stop codon (1383 bases), and a 3` untranslated region (1390 bases) (fig 4B).20 The open reading frame encodes a pre-pro-protein in which the pre-sequence (or signal sequence) directs factor IX for secretion, the pro-sequence provides a binding domain for a vitamin K dependent carboxylase, which carboxylates certain glutamic acid residues in the adjacent GLA domain, and the remainder represents the factor IX zymogen, which enters the circulation after removal of the pre- and pro-sequences. Domains within the zymogen are identified according to structure or function.
Exon and intron sizes for the human factor IX gene2
(A) Genomic organisation of the human factor IX gene. Exon and intron sizes are given in table 2. (B) Factor IX mRNA showing the relative size and location of the open reading frame. (C) The newly synthesised factor IX protein molecule comprising a pre- and pro-sequence (27 and 19 amino acids, respectively) and a mature peptide of 415 amino acids (total length, 461 amino acids). (D) Activated factor IX comprising an N-terminal light chain and a C-terminal heavy chain held together by a disulphide bridge between cysteine resides 132 and 279. GLA, “GLA” domain, in which 12 glutamic acid residues undergo post-translational γ-carboxylation by a vitamin K dependent carboxylase; EGF, epidermal growth factor-like domain; act, activation peptide released after proteolytic activation at arginine 145 and arginine 180; catalytic, the serine protease domain.
Activation of factor IX involves cleavage of two peptide bonds, one on the C-terminal side of arginine 145 (the α-cleavage) the other on the C-terminal side of arginine 180 (the β-cleavage) (fig 4C).21 These cleavages are brought about by activated factor XI generated through the intrinsic pathway,22 or via the tissue factor/activated factor VII complex of the extrinsic pathway.23,24 The relative importance of these two pathways to factor IX activation is indicated by the fact that severe factor XI deficiency is typically associated with a mild bleeding disorder, whereas severe factor VII deficiency is characterised by a severe bleeding disorder. The activation cleavages generate an N-terminal light chain and a C-terminal heavy chain, held together by a disulphide bond between cysteine residues 132 and 279 (fig 4D).21
The above overview is necessarily brief. Its purpose is to relate salient features that provide the context and framework for the molecular information that follows. More in depth coverage can be obtained from several excellent reviews.14,25–28
POLYMORPHISMS
The factor VIII and factor IX genes both contain two types of polymorphism: single nucleotide polymorphisms (SNPs) and length polymorphisms, also known as variable number tandem repeat sequences (VNTRs) or microsatellites. Polymorphisms have a scientific interest of their own and are useful in areas as diverse as forensic science29,30 and the study of human evolution.31,32 They have clinical relevance in the context of hereditary disorders in that they can be used to track a defective (or normal) gene through an affected family. Such linkage studies have permitted carrier status investigation and prenatal diagnosis in haemophilia A and haemophilia B.33,34
There is an apparent paucity of polymorphisms in both the factor VIII and factor IX genes. In the recent presentation of the sequence of the human genome,35 two haploid human genomes chosen randomly differed by 1 base/1250 bases on average; therefore, the factor VIII and factor IX genes would be expected to contain approximately 144 and 27 SNPs, respectively. However, these numbers are considerably greater than the number of polymorphisms detected so far in these genes, although this could reflect a problem of detection rather than a genuine paucity. Certainly, many more candidate polymorphisms have been identified as a result of the human genome sequencing programme35,36 than the recognised polymorphisms described below.
“There is an apparent paucity of polymorphisms in both the factor VIII and factor IX genes”
FACTOR VIII GENE POLYMORPHISMS
The factor VIII gene contains several SNPs, many of which fall into the subcategory of restriction fragment length polymorphisms (RFLPs) (fig 5A). In addition, two microsatellites have been reported within the gene, one in intron 13,38 the other in intron 22 (fig 5A).44

Some of the known polymorphisms in the human genes for (A) factor VIII and (B) factor IX. Source references: factor VIII gene intron 7 G/A,37 intron 13 (CA)n,38 intron 18 BclI,39 intron 19 HindIII,40 intron 22 XbaI A,41,42 intron 22 MspI A,43 intron 22 (CA)n,44 intron 25 BglI,45 3` MspI,46 factor IX gene 5` −793 G/A,47 5` BamHI,48 5` MseI,49 intron 1 DdeI,50 intron 3 XmnI,50 intron 3 BamHI,51 intron 4 TaqI,52 intron 4 MspI,53 exon 6 MnlI,54 exon 8 (RY)n,55 and 3` HhaI.56
The intron 13 microsatellite comprises the simple repeat (CA)n,38 whereas the intron 22 comprises (CA)n(CT)n.44 Differences in the value of “n” at such repeats have been attributed to DNA polymerase slippage and appear to drift towards an increase in repeat length.57 Because such events could occur between generations within a family, microsatellite data should be interpreted with appropriate caution in a linkage study.58 Preferably, verification of microsatellite data should be sought from at least one other intragenic locus.
The XbaI and MspI RFLPs in intron 22 of the factor VIII gene (designated XbaI A and MspI A) occur within the int22h-1 sequence.10,42,43 The corresponding positions in the extragenic homologues int22h-2/3 are also polymorphic for XbaI42 and MspI43 (designated XbaI B and C, and MspI B and C, respectively). Caution must therefore be exercised in the use of these two highly informative loci.
The allele frequencies of some of the polymorphisms have been investigated in various populations and differences are seen (table 3). Three observations are of particular interest: first, the [+] alleles for the BclI and HindIII RFLPs have very different frequencies in American blacks compared with other ethnic groups, affording a small glimpse into human evolutionary divergence. Second, although whites and Chinese have similar allele frequencies at the BclI, HindIII, and XbaI A loci, they differ noticeably at the BglI locus. The BglI RFLP is the result of a transition that is not likely to recur independently.62 Thus, it is possible that its presence in whites but not in Chinese might reflect its emergence in the former after the two ethnic groups diverged. However, the presence of the BglI RFLP in Japanese and American blacks suggests that this explanation is too simplistic. Third, in American blacks the allele frequencies for the BclI and HindIII loci are the reverse of what is seen in other ethnic groups; however, for the BglI locus, this is not the case. This complicates the interpretation of the ancestral factor VIII gene haplotype(s) from which the haplotypes existing in present day populations must have evolved.
Allele frequencies for four factor VIII gene polymorphisms frequently used in linkage studies in haemophilia A
The differences in allele frequencies between populations have implications for linkage studies in haemophilia A: the loci of greatest diagnostic potential may differ according to the ethnicity of the family to be investigated and this has to be borne in mind in assessing the investigative strategy.
Linkage disequilibrium has not been investigated for all loci, although it has been shown for some. There is strong linkage disequilibrium between the intron 18 BclI, intron 7 G/A, intron 19 HindIII, and intron 25 BglI RFLPs.59,63,64 The linkage disequilibrium in these cases is strong but not complete; therefore, additional informativity might be obtained from their inclusion in a linkage study. The intron 18 BclI and intron 22 XbaI A loci also show linkage disequilibrium41; however, this is less pronounced and their combined analysis provides much more informativity than either used on its own. For example, in whites, the informativity for combined use v individual use of XbaI A is approximately 65% v 49%,41 in Chinese it is 52% v 49%,61 and in Japanese it is 79% v 48%.60
XbaI A and MspI A are located within 737 bases of each other.43 Despite their close proximity, the RFLPs are not in complete linkage disequilibrium. Haplotype analysis from a white population predicts an informativity of approximately 60% in linkage studies using both, compared with 47% using either on its own.43
Linkage disequilibrium, like allele frequencies, differs between ethnic groups and this should also be taken into account in assessing the linkage analysis strategy within a family.34 Linkage disequilibrium and low rates of heterozgosity decrease the overall potential informativity of the polymorphisms in the factor VIII gene such that, in practice, about 10–15% of linkage studies are non-informative. The addition of further polymorphic loci to the small array already available would therefore be valuable.
FACTOR IX GENE POLYMORPHISMS
The human factor IX gene contains several SNPs, most of which fortuitously fall into the subcategory of RFLPs (fig 5B). The MnlI polymorphism54 is located in exon 6 and brings about the amino acid substitution threonine/alanine at position 148 (known as the Malmö polymorphism65). Therefore, the factor IX protein itself is polymorphic and the Malmö polymorphism can be genotyped at the protein level using antibodies specific for each variant.66,67 There are two microsatellite loci, one in intron 168 (the “DdeI” polymorphism50) and one in the 3` untranslated region of exon 855 (fig 5B). These are of the type (RY)n (R, purine; Y, pyrimidine).
As seen for polymorphisms in the factor VIII gene, factor IX gene polymorphisms show ethnic variation, which in some instances is quite pronounced (table 4). For example, the allele frequencies of the BamHI RFLPs in the 5` region and in intron 3 are reasonable in the American black population, whereas these loci are essentially non-polymorphic in whites, Chinese, and Japanese (table 4). In contrast, the MnlI RFLP in exon 6 is common in whites and American blacks, but rare in Chinese (table 4). These differences in allele frequencies between racial groups impact upon linkage studies and influence which loci should be included for familial investigation in haemophilia B.34
Allele frequencies for several factor IX gene polymorphisms in different ethnic groups
The (RY)n microsatellites have been investigated in considerable depth.55,68,73 The 3` (exon 8) locus has the consensus (GT)a(ATGCGT)4AG(AC)bGCAT(AC)3(AT)2 and four alleles have been demonstrated (I–IV) in which the values of “a” and “b” vary.55 The intron 1 (RY)n motif comprises two units, A and B, with consensus sequence GT(AC)3(AT)3(GT(AT)I (I = 4 for the A unit and 5 for the B unit).68 The polymorphism is caused by variability in the number of A and B units present. Both microsatellites show variation between racial groups.55,73 Unlike the microsatellites within the factor VIII gene, the variability in the microsatellites of the factor IX gene does not appear to arise as a result of replication slippage. Instead, the patterns of variation observed are compatible with sister chromatid exchange (homologous recombination).68
There is strong circumstantial evidence that (RY)n sequences are involved in homologous recombination.68 In this light, it is interesting to note that the BamHI RFLP which is 5` to the intron 1 (RY)n is in linkage equilibrium with polymorphisms located between this (RY)n and the exon 8 (RY)n.68 Similarly, the HhaI RFLP 3` to the exon 8 (RY)n is in linkage equilibrium with polymorphisms between this (RY)n and the intron 1 (RY)n.68 In contrast, the polymorphisms between the intron 1 and exon 8 (RY)n are in strong linkage disequilibrium.26,50,72
If the (RY)n sequences of the factor IX gene truly are involved in homologous recombination events, this has evident implications for linkage studies (the loss of linkage between polymorphic loci and the defect in the factor IX gene). However, this has not been seen, although the possibility of its occurrence argues for the inclusion of several loci across the factor IX gene in a linkage study.
Linkage disequilibrium has been investigated for some of the factor IX gene polymorphisms. In whites, the TaqI, XmnI, MspI, and MnlI RFLPs show pronounced linkage disequilibrium, such that the use of all four loci would only increase informativity to 55% compared with 45% when using the TaqI site alone.26,50,72 The DdeI and HhaI polymorphisms, however, show much less disequilibrium,72 possibly for the reasons discussed above.
In American blacks, the TaqI, MspI, and intron 3 BamHI loci show linkage disequilibrium51; however, the 5` BamHI, intron 1 (RY)n, and XmnI polymorphisms appear to be in equilibrium.51,72,73 Once again, these observations may be explained by the involvement of (RY)n sequences in homologous recombination.
“As seen for polymorphisms in the factor VIII gene, factor IX gene polymorphisms show ethnic variation, which in some instances is quite pronounced”
The G/A SNP at −793 is only 104 bases from the MseI RFLP at −698.49 Loci in such close proximity would be expected to show complete linkage disequilibrium; however, this is not the case. Their combined use provides an informativity of 60% compared with approximately 45% for either marker used alone.49
No linkage disequilibrium has been seen between the 3` HhaI RFLP and other intragenic loci. Taken together with the possible loss of linkage between loci either side of the (RY)n sequences, a theoretical minimum linkage study with high diagnostic potential can be proposed. This would use HhaI 3` to the exon 8 (RY)n, a second polymorphism 5` to the intron 1 (RY)n, and a third polymorphism between the (RY)n loci. The second and third loci should be chosen according to racial background.
LINKAGE ANALYSIS IN HAEMOPHILIA A AND B
In essence, the practical approach involves the extraction of genomic DNA from anticoagulated whole blood obtained from relevant family members, amplification of DNA spanning the target polymorphic locus using the polymerase chain reaction (PCR), and then analysis of the PCR product using any of a variety of techniques to distinguish the alleles present75 (for example, restriction digestion, single strand conformation polymorphism analysis, heteroduplex analysis,64 PCR product length analysis).
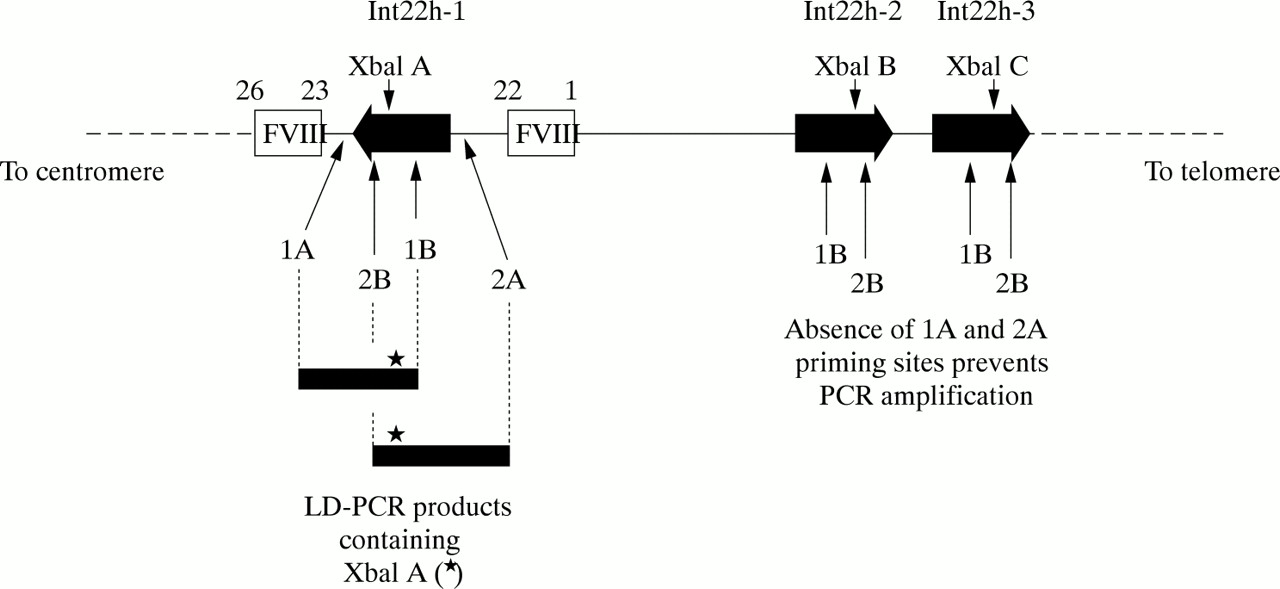
For most of the polymorphic loci in both genes this is a straightforward task and is the mainstream approach in linkage analyses.34,75 However, for the XbaI A and MspI A RFLPs in intron 22 of the factor VIII gene, the task is complicated by the extragenic homologues (XbaI B and C, and MspI B and C). Until recently, specific amplification of these loci in int22h-1 was not possible. However, the introduction of long distance PCR (LD-PCR), in which stretches of genomic DNA as long as tens of kilobases can be amplified from small amounts of starting material, paved the way for the solution of specific amplification of the int22h-1 loci.76,77
The approach is outlined in fig 6. One LD-PCR primer hybridises to all three int22 homologues, whereas a second primer hybridises only to the factor VIII gene sequence outside the int22h-1 region. In the LD-PCR, exponential amplification is only possible for the int22h-1 target as a result of the hybridisation of both primers; int22h-2 and int22h-3 fail to amplify because only one primer can hybridise.
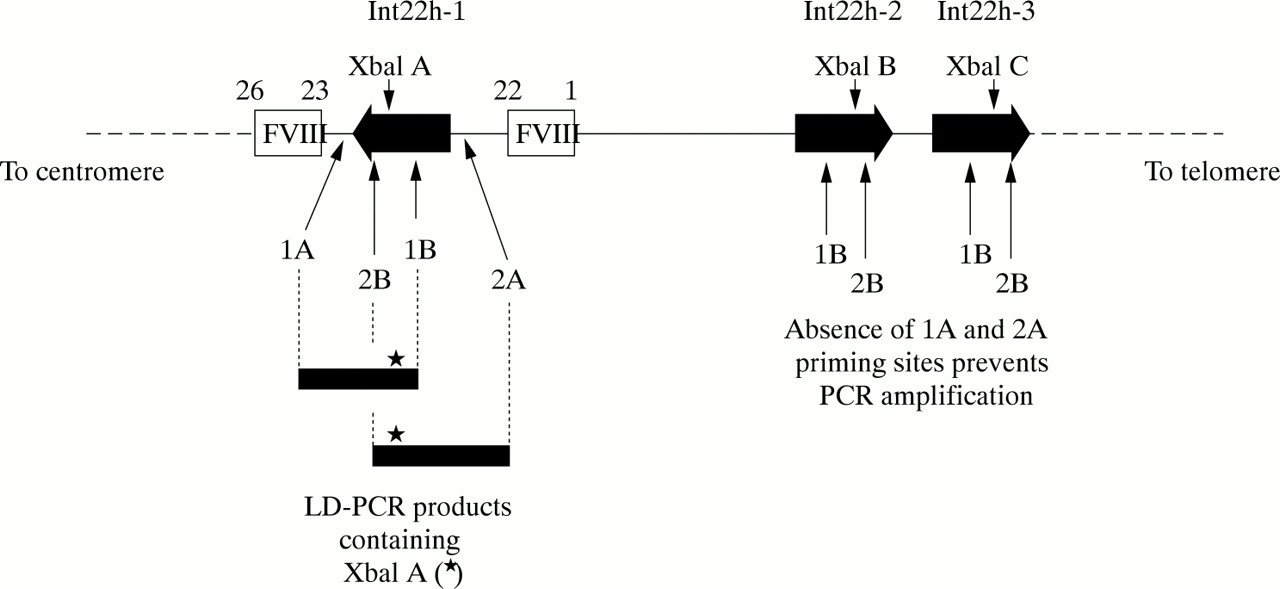
Genotyping the intron 22 XbaI A restriction fragment length polymorphism (RFLP) using long distance PCR (LD-PCR). A primer specific for factor VIII gene sequence (1A or 2A) is used in conjunction with a primer that flanks the XbaI A site and that hybridises within the int22h homologue (1B or 2B, respectively). The resulting LD-PCR product (∼ 7 kb) contains XbaI A (asterisk) and can be genotyped by XbaI digestion. The analysis is specific for XbaI A because the sequences to which primers 1A or 2A hybridise do not occur at the extragenic int22h loci. Primers 1B or 2B can hybridise, but in the absence of primers 1A and 2A amplification cannot take place.
The resulting LD-PCR product can then be genotyped for XbaI A by digestion with this restriction endonuclease,76,77 or it can be used as the substrate in a second round of PCR that contains primers for amplification of the MspI A site also contained within the LD-PCR product. MspI A can then be genotyped by digestion of the second round product with MspI.77
It is clear that PCR has provided a major step forward for the analysis of polymorphisms in the haemophilias. The traditional approach for RFLP analysis, using Southern blot, was labour intensive, time consuming, hazardous, and required relatively large quantities of high molecular weight DNA. In comparison, PCR is rapid, non-labourious, non-hazardous, and requires only small quantities of DNA, which can be degraded to a considerable degree.
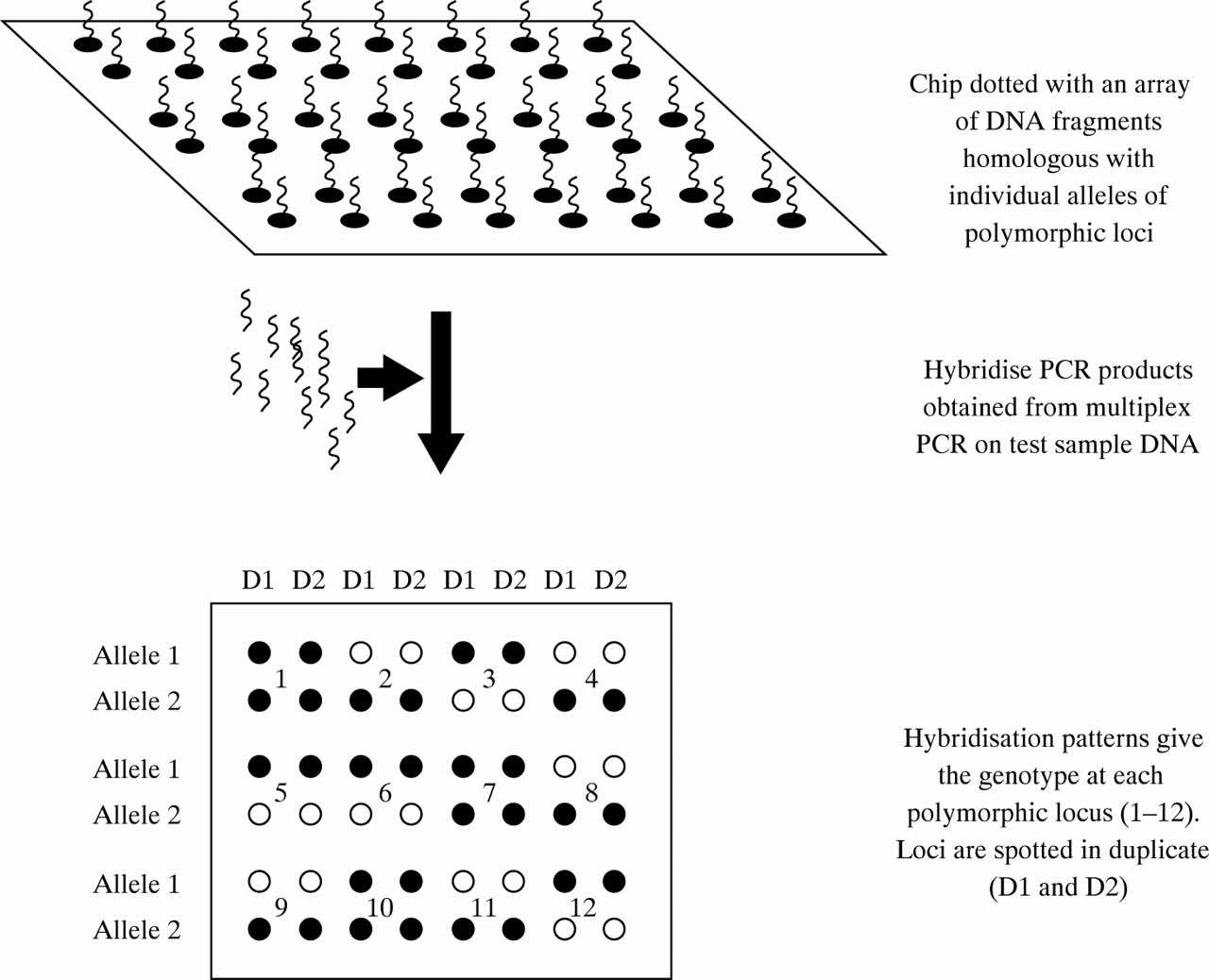
Therefore, it is not surprising that the use of PCR is now common to most, if not all, laboratories engaged in genetic testing in the haemophilias. However, the methods used for post-PCR product analysis differ enormously. At one end of the spectrum there are low tech approaches, such as agarose gel electrophoresis followed by ethidium bromide staining, whereas at the other there are the high tech approaches such as the Light CyclerTM or fluorescence based capillary electrophoresis. The choice within laboratories reflects many parameters that need not concern us here; the important point is that post-PCR product analysis is flexible and amenable to the most up to date technologies in molecular biology. Whether “DNA chips”78–80 (fig 7) may one day become useful for linkage studies in the haemophilias remains to be seen: the evolving movement towards direct mutation detection covered in the next section may ultimately result in linkage analysis being pushed into the sidelines.

One approach to polymorphism screening using DNA chips. Multiplex PCR could be used to generate locus specific fragments for hybridisation to the chip or, conceivably, fragmented, labelled genomic DNA could be hybridised directly, without amplification.
THE MOLECULAR BASIS OF THE HAEMOPHILIAS
The mutations causing haemophilia A and B have been localised and characterised in several hundreds of patients. What is immediately evident from the enormous number of mutations that have been elucidated is that the molecular basis of the haemophilias is extremely diverse. This review will focus on some of the principal generalisations that emerge from an inspection of the vast databases of haemophilia mutations and it will consider some specific mutations that illustrate the differing molecular pathologies that exist.
The relation between cause and effect is not always clear, and only in a minority of mutations do we have a good understanding of how haemophilia ensues from the gene defect that is present. Unfortunately, this is mostly confined to those mutations whose effect is self evident in that the gene is disrupted, translation is prematurely terminated, activation of the protein is hindered, or mRNA splicing is corrupted, etc. For most mutations, there is no obvious explanation for effect, in which case one is left with candidate explanations such as protein destabilisation, incorrect folding, perturbation of structurally or functionally important regions, and so on. These are often difficult possibilities to prove. However, x ray crystallography and advances in software for molecular modelling and prediction of protein structure have permitted these possibilities to be explored for some mutations.81–84
OVERVIEW OF MUTATIONS
Mutations that have been characterised in the haemophilias are listed in the following databases on the World Wide Web (these databases are freely accessible).
-
Haemophilia A: http://europium.csc.mrc.ac.uk/usr/WWW/WebPages/main.dir/main.htm (HAMSTeRS, The Haemophilia A Mutation, Structure, Test, Resource Site).
-
Haemophilia B: http://www.umds.ac.uk/molgen/ (Haemophilia B Mutations Database).
-
Both can be accessed via: http://archive.uwcm.ac.uk/uwcm/mg/hgmd0.html (Human Gene Mutation Database).
The databases are extensive listings that represent a vast coverage both of published and unpublished findings.85 From these listings, several generalisations emerge that are common to both haemophilia A and B. The following is a synthesis of those generalisations.
TYPES OF MUTATION
Point mutations, deletions, insertions, and rearrangements/inversions are found in both the factor VIII and factor IX genes. Point mutations (single nucleotide substitutions) are the most common gene defect and are present in approximately 90% of patients. Deletions are the second most common gene defect and are present in approximately 5–10% of patients. Insertions and rearrangements/inversions are quite rare within the haemophilia population, with the exception of an inversion that is prevalent among patients with severe haemophilia A (this inversion, the intron 22 inversion, will be discussed in more detail later).
Point mutations
The point mutations that occur in the haemophilias comprise missense mutations (these change a codon so that a different amino acid is encoded), nonsense point mutations (these change an amino acid codon into a translation stop codon), and mRNA splice site point mutations (these corrupt a true mRNA splice site, or create a novel one).
Missense mutations
The severity of haemophilia arising from missense point mutations depends very much on the nature of the amino acid substitution and its location. Semiconservative amino acid substitutions (the new amino acid is similar to the normal one either with respect to charge, hydrophobicity, polarity, or shape) tend to be associated with less severe disease except when they occur in regions of structural or functional importance. Non-conservative amino acid substitutions (the new amino acid is very dissimilar to the normal amino acid) tend to be associated with moderate to severe disease wherever they occur in the protein. The scope for structural perturbation/destabilisation is greater for non-conservative amino acid substitutions; therefore, there is a higher probability of their having a more detrimental overall effect.
Nonsense mutations
The outcome is expected to be a truncated protein molecule. This may, or may not, get as far as entering the circulation, but even if it does, truncation is highly likely to have made it defunct. It is therefore not surprising that nonsense mutations are associated with severe haemophilia. Exon skipping is a further possibility arising from a nonsense mutation86,87 and this is also extremely detrimental: an in frame skip will result in a protein lacking the amino acids encoded by the skipped exon, an out of frame skip will result in a frame shift.
mRNA splice site mutations
Mutations that destroy or create mRNA splice sites are associated with variable severity of haemophilia; this very much depends on whether some correct transcripts can be processed (mild to moderate disease) or whether there is a complete loss of correct mRNA processing (severe disease). Exon skipping is a further possible consequence of splice site mutations, the outcome of which depends on whether the skip is in frame or results in a frame shift.88
Point mutations can be grouped according to whether or not they are classic CG site transitions. CG sites are hypermutable and give rise to C to T transitions or G to A transitions.89 The mechanism underlying these transitions is understood89 and is a major mechanism of human gene mutation. This is clearly exemplified in the haemophilias, in which approximately 30% of all distinct point mutations arise at CG sites. The remaining 70% of distinct point mutations do not occur at CG sites and may arise—for example, as a result of nucleotide misincorporation during DNA replication.
Point mutations may also be grouped according to whether or not they are recurrent. Recurrent mutations occur in both haemophilia A and B and are principally found at CG sites. The hypermutability of such sites underlies the independent recurrence of their transition events. There are rare examples of recurrent mutations that do not occur at CG sites. These are generally explained by a founder event that has multiplied in successive generations. Evidence for this may include confined geographical distribution and identical polymorphic haplotype in all affected genes, irrespective of geographical distribution.90,91 Founder mutations typically cause mild/moderate disease; a mutation causing severe disease has a low probability of survival over many generations.
Deletions
Deletions of the factor VIII and factor IX gene include whole gene deletions, partial gene deletions at the 5` end, 3` end, or within the gene, and microdeletions of one to several base pairs (gross deletions are compiled in several reviews14,28,92,93). They do not appear to cluster to a specific region of either gene, but appear to be randomly distributed. A deletion has a high probability of destroying genetic function, removing a chunk of protein, or introducing a frame shift, all of which are extremely detrimental. Not surprisingly therefore, deletions are generally associated with severe disease.
Insertions
Insertions have been characterised in both the factor VIII and factor IX genes. Like deletions, these can be gross or as small as one or several nucleotides, and gene function or the gene product can be very adversely affected (gross insertions are compiled in several reviews14,28,92,93).
“Deletions are generally associated with severe disease”
Rearrangements/inversions
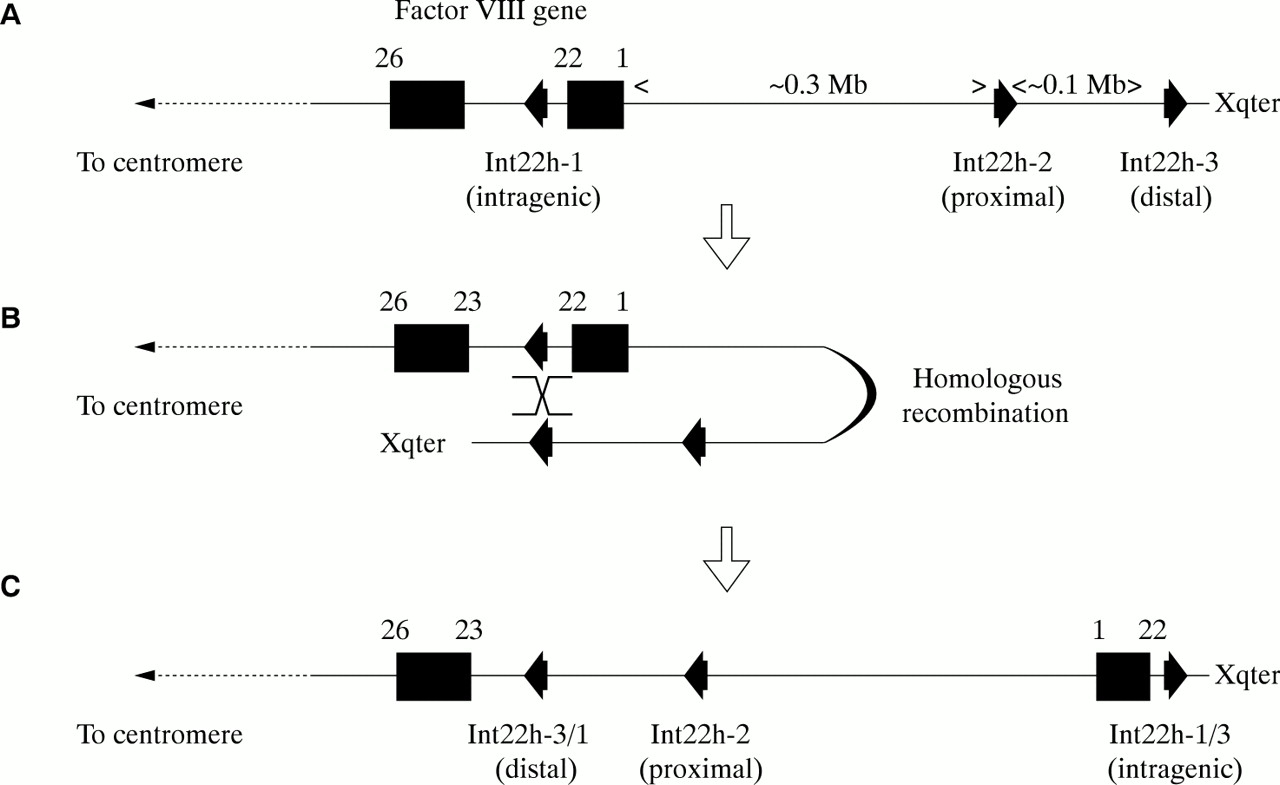
These are rare gene defects in the haemophilias, except for an inversion involving intron 22 of the factor VIII gene, which causes severe haemophilia A, and which is found in 40–50% of patients with severe disease.94,95
The intron 22 inversion arises through homologous recombination between int22h-1 and int22h-2 (proximal) or int22h-3 (distal) during meiosis94,95 (fig 8A–C). After homologous alignment, recombination results in the 5` portion of the factor VIII gene being juxtaposed to the telomeric DNA, whereas the relevant extragenic homologue becomes juxtaposed to the 3` portion of the factor VIII gene (fig 8B). In the re-linearised chromosome, the result is an inversion of the sequence between int22h-1 and int22h-2 or int22h-3, depending on which extragenic copy aligned (fig 8C). The factor VIII gene is completely disrupted: introns 1 to 22 are moved away from their normal context and their orientation is inverted. Severe haemophilia A results.

The intron 22 inversion of the factor VIII gene. (A) The normal configuration of the factor VIII gene and int22h homologues. (B) The distal or the proximal int22h homologue aligns with the intragenic int22h homologue (distal shown here). Homologous recombination takes place. (C) Linearisation of the X chromosome gives a completely disrupted factor VIII gene: exons 1 to 22 are displaced towards the telomere and are orientated in a direction opposite to their normal orientation. A contiguous factor VIII gene transcript running from exon 1 to 26 is no longer possible and severe haemophilia A results.
The intron 22 inversion is principally an error of DNA replication during spermatogenesis and not oogenesis96: in males, the absence of a second X chromosome for homologous alignment during meiosis would favour intrachromosomal alignments where they are possible. Thus, in haemophilia A families in which the intron 22 inversion is the causative gene defect, the defect can often be shown to have originated in an unaffected male relative, and in sporadic cases, this is often the patient's grandfather on his mother's side of the family.96 The distal inversion is more common than the proximal inversion,96 which may be explained by the greater genetic distance between the factor VIII gene and the distal int22h-3 homologue facilitating the formation of the loop required for alignment to take place.
The intron 22 inversion was, until recently, diagnosed using the labour intensive approach of Southern blot analysis (the DNA fragmentation pattern obtained for the normal gene differs from that obtained for an inverted gene, and the patterns for the distal and proximal inversions also differ).94,95 This has now been superseded by an approach using LD-PCR (fig 9).97 This impressive strategy detects normal and inverted genes; however, it does not distinguish proximal inversion from distal inversion.

Detection of the intron 22 inversion of the factor VIII gene using long distance PCR (LD-PCR).97 Primers P and Q hybridise specifically to sequences within the factor VIII gene and flank int22h-1. Primers A and B hybridise specifically to sequences flanking int22h-2 and int22h-3. The normal factor VIII gene therefore gives the LD-PCR products P + Q (12 kb) and A + B (10 kb). The intron 22 inversion results in the hybridisation site for primer Q being displaced towards the telomere, whereas the hybridisation site for primer B from the distal or proximal int22h homologue (depending on which was involved in the homologous recombination leading to the inversion, distal shown here) is brought into context with primer P. The inverted gene therefore gives the LD-PCR products P + B (11 kb), A + B (10 kb), and Q + A (11 kb). An A + B product is always obtained because one copy of the int22h homologues is unaffected by the inversion.
Although most intron 22 inversions involve int22h-1 and 2/3, atypical inversions have been reported.94,98,99 Detailed analysis of one of these revealed a third, truncated copy of int22h between the factor VIII gene and int22h-2, and this was involved in the inversion.98 These atypical inversions give anomalous patterns using Southern blot. They may fail to be detected using LD-PCR and this has implications for diagnosis using this approach. Fortunately, they appear to be rare.
MOLECULAR PATHOLOGY: CAUSE–EFFECT RELATIONS
The following detailed examples offer a glimpse into the many and varied mechanisms through which mutations bring about their effects. Mutations can exert their effects at any stage from genetic function right through to protein function, and often they reveal important properties of these functions that might otherwise have escaped realisation. The investigation of cause–effect relations is therefore of considerable value with potentially high rewards.
Nonsense mutations at CGA (arginine) codons
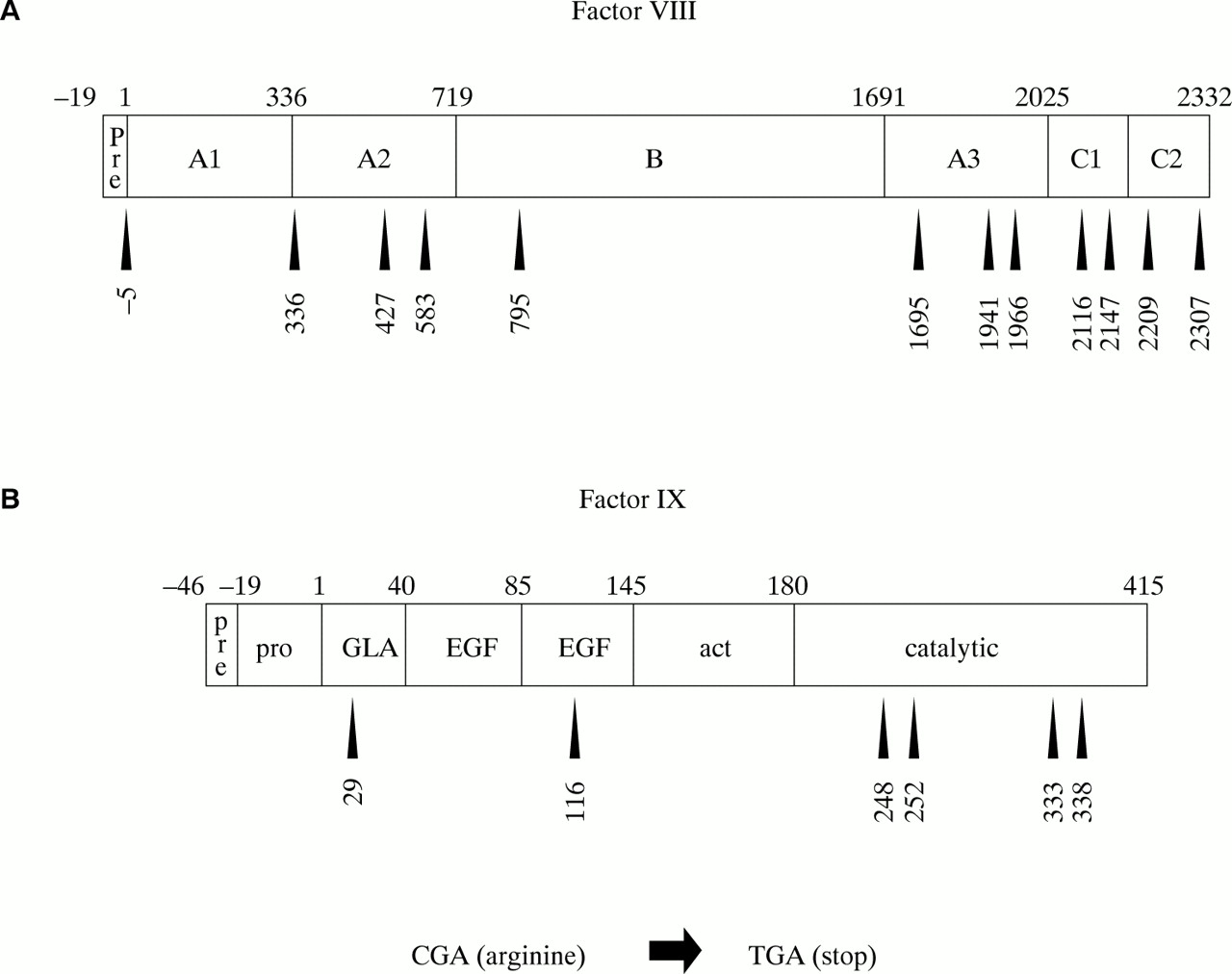
CG transitions can give rise to missense or nonsense mutations, depending upon the codon in which the CG site resides and whether C to T or G to A transition has occurred. C to T transition is particularly damaging at arginine codons of the type CGA: the result is the translation stop codon TGA (a nonsense mutation). As discussed above, nonsense mutations are particularly detrimental and are associated with severe disease. The factor VIII open reading frame has 70 CG sites, 12 of which reside in arginine codons of the type CGA (fig 10A). The factor IX open reading frame has 20 CG sites, six of which reside in CGA codons (fig 10B). C to T transition resulting in nonsense mutations has been reported at all of these codons and is associated with severe haemophilia, either haemophilia A100–104 or B105–107. As would be expected from the hypermutability of CG sites, many of these nonsense mutations are recurrent among patients with haemophilia.
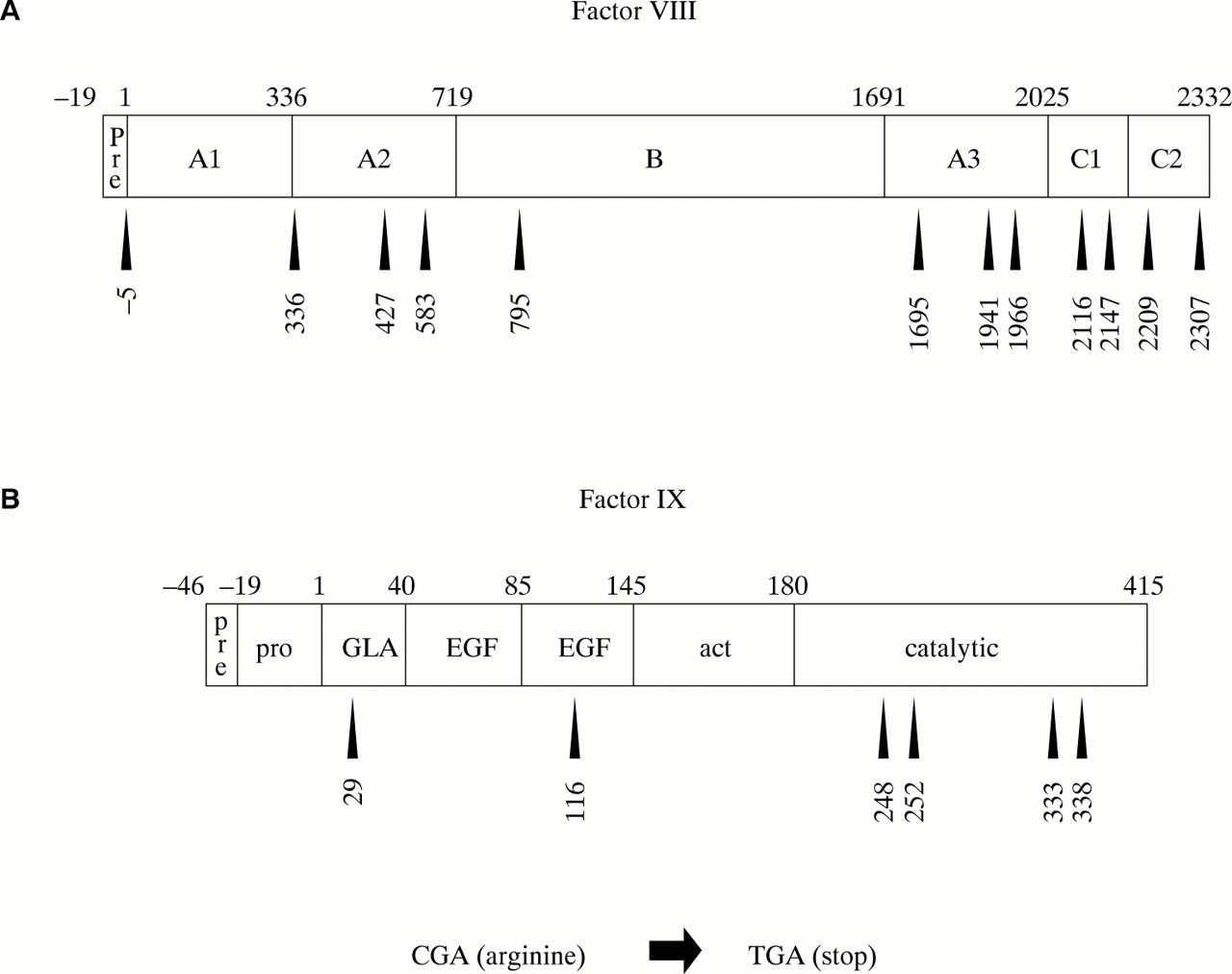
Arginine residues encoded by CGA within (A) the factor VIII and (B) the factor IX protein sequences. Nonsense mutations are brought about by C to T transition within the CG site of these codons. These are recurrent gene defects within haemophilia populations.
Missense mutations at activation cleavage sites
The proteolytic cleavages that activate factor VIII and factor IX occur after arginine residues located at positions 372 and 1689 in factor VIII (fig 3C), and 145 and 180 in factor IX (fig 4C). The codons for these arginine residues all contain the dinucleotide CG; transition of C to T or G to A within any of these codons causes haemophilia, either haemophilia A103,104,108 or B.107,109,110 Either transition results in a missense mutation that causes the relevant activation cleavage site arginine to be replaced by another amino acid (fig 11A, B). This compromises the activation cleavage at that site. Normal activation can therefore take place at only one of the two necessary sites and the result is a factor VIII or factor IX molecule with decreased function. Partial activity may remain, depending upon the activation cleavage site that is lost and the amino acid substitution. For example, factor IX cleaved at the β cleavage site alone retains partial activity, whereas factor IX cleaved at the α cleavage site alone is inactive; β cleavage is essential for unmasking the serine protease function of the catalytic domain. Thus, mutant proteins such as factor IX Chapel Hill111 (histidine 145) retain some residual activity upon cleavage at the β bond, despite loss of cleavage at the α bond. The same is true for the cysteine 145 mutation (factor IX Albuquerque112). Mild haemophilia B results. In contrast, factor IX Hilo113 (glutamine 180) cannot be activated at the β cleavage site, which is essential for the function of the enzyme, and severe haemophilia B results.
Mutations as a result of CG transitions at the codons encoding the activation cleavage site arginine residues of (A) factor VIII and (B) factor IX.
Mutations affecting factor VIII binding to von Willebrand factor
In plasma, factor VIII circulates bound to von Willebrand factor and this interaction stabilises factor VIII.16,17 The regions of factor VIII that were found to be important for this interaction are the N-terminal part of the A3 domain, the C2 domain and, more recently, the C1 domain.114–117 Mutations that interfere with factor VIII binding to von Willebrand factor illustrate how different mechanisms can affect the same property.
Within the A3 domain, sulphation of tyrosine 1680 is crucial for factor VIII interaction with von Willebrand factor.118 Missense mutations resulting in the replacement of this tyrosine residue result in decreased factor VIII binding to von Willebrand factor104,118,119 Similarly, retention of the tyrosine residue, but prevention of sulphation, also decreases factor VIII binding to von Willebrand factor.120 Clearly, the loss of sulphation is crucial, whether or not this is accompanied by structural/folding changes brought about by a mutant amino acid substitution.
Several mutations have been identified within the C1 domain that decrease factor VIII binding to von Willebrand factor.117,121 Some of these occur in residues that are conserved in the discoidin family of proteins; therefore, they are more likely to play a structural role in the C1 domain than to be involved directly in intermolecular interactions with von Willebrand factor. The structural perturbation that ensues compromises the interaction between the two proteins. Examples of such mutations are serine 2119 to tyrosine, asparagine 2129 to serine, arginine 2150 to histidine, and proline 2153 to glutamine.117 However, none of these mutations sheds light on the mechanism by which C1 influences the affinity of factor VIII for von Willebrand factor: residues within the C1 domain may directly interact with von Willebrand factor residues; alternatively, mutations in the C1 domain may have an indirect effect through perturbation of folding or structure.
Besides mutations within the factor VIII gene that affect interaction with von Willebrand factor, there are also mutations of the von Willebrand factor gene that decrease the affinity of von Willebrand factor for factor VIII (von Willebrand disease type 2N).122 Von Willebrand disease type 2N is typified by decreased factor VIII values, but shows an autosomal pattern of inheritance (the von Willebrand factor gene is located on chromosome 12 at 12p12–12pter). For obvious reasons, von Willebrand disease type 2N has also been referred to as pseudohaemophilia A.123 There is an increasing list of mutations underlying von Willebrand disease type 2N. These are listed on the World Wide Web at URL http://mmg2.im.med.umich.edu/vWF/ (von Willebrand Factor (VWF) Database).
Mutations affecting factor VIII secretion
Factor VIII values may be decreased in the circulation by mutations that compromise its secretion. The haemophilia A missense mutations arginine 593 to cysteine and asparagine 618 to serine cause intracellular accumulation of factor VIII when expressed in cell culture.124 This was concurrent with decreased factor VIII activity and antigen in the conditioned medium of transfected cells. Intracellular accumulation and decreased secretion of factor VIII possessing either of these mutations may explain the reduction of both factor VIII activity and antigen in plasma of patients with mild haemophilia A and these gene defects.
Mutations affecting factor IX binding to factor VIII
Initial evidence indicating that the protease domain of activated factor IX is involved in binding to activated factor VIII was provided by the inhibition of the interaction by a monoclonal antibody directed against the protease domain.125 Subsequent antibody and mutation studies have revealed a complex story.
The factor IX protease domain contains a Ca2+ binding site in which glutamic acid residue 245 participates in binding the metal ion.126 Substitution of glutamic acid 245 by valine (a haemophilia B mutation) results in a factor IX molecule that is rapidly cleaved between arginine 318 and serine 319 by activated factor XI.127 This variant factor IX has a 15 times lower affinity for factor VIII before cleavage at arginine 318 and a 120 times lower affinity after cleavage.127 Currently, it is believed that binding of Ca2+ to the protease domain protects factor IX from proteolysis after arginine 318, and this stabilises a neighbouring surface exposed helix (residues 330–338), which binds factor VIII.84,127
Mutations of eight of the nine residues in this helix (leucine 330 to arginine 338) are reported in the haemophilia B population and they probably cause haemophilia, at least in part, by directly interfering with factor IX–factor VIII interaction. In contrast, the haemophilia B mutation glutamic acid 245 to valine abolishes Ca2+ binding and this has an indirect effect on factor VIII binding, through destabilisation and possible structural effects, as discussed above.128
Mutations causing factor IX deficiency during anticoagulant treatment
Bleeding complications arising from oral anticoagulant treatment are not uncommon. An unusually selective decrease in factor IX procoagulant activity (1–3% of normal) has been observed in some patients receiving coumarin.129 Normal factor IX activity was recovered on withdrawal of the anticoagulant. Two different missense mutations at alanine −10 were found to be implicated: alanine to valine and alanine to threonine. Alanine −10 lies within the pro-peptide domain, at a position that is essential for binding of the carboxylase that modifies glutamic acid residues in the adjacent GLA domain (fig 4C).130,131 This carboxylase requires vitamin K as cofactor.132,133 Coumarin derivatives interrupt the vitamin K cycle and lead to rapid depletion of the cofactor form of the vitamin.134 Carboxylase activity is consequently decreased and this, in conjunction with a decreased affinity of the carboxylase for the mutant pro-domain,131,135 results in the loss of GLA carboxylation and hence of factor IX activity. Presumably, in the absence of coumarin, normal vitamin K concentrations supply the carboxylase adequately and this permits appropriate processing of the GLA domain, even though the affinity of the carboxylase for the pro-domain may be decreased by the alanine −10 mutations.
Mutations in the factor IX promoter and haemophilia B Leyden
The mutations discussed so far have affected either the factor VIII or factor IX coding sequence, with a consequent impact upon the translated protein. However, certain mutations within the promoter of the factor IX gene do not result in a qualitative change in the factor IX molecule. These mutations lie within sequences to which proteins involved in transcription bind. They disrupt or interfere with protein–DNA interactions in the promoter and so compromise the efficiency of transcription.
Some promoter mutations cause haemophilia B that is present throughout life.136 However, others cause a variant of haemophilia B known as haemophilia B Leyden, which is characterised by severe factor IX deficiency at birth and through childhood, an increase in factor IX values from puberty through adolescence, and near normal factor IX values throughout adulthood.137
The molecular pathology of haemophilia B Leyden has been investigated intensively. The promoter of the factor IX gene contains a binding site for the CCAAT/enhancer binding protein138 (C/EBP) and for the rat liver protein LF-A1/HNF4 (fig 12).136 These proteins are involved in transcription; therefore, the binding sites may act as constitutive promoter elements for gene expression. The promoter also contains a candidate androgen response element (ARE), which overlaps the LF-A1/HNF4 element (fig 12).136,139 This may act as a hormonally regulated promoter element for gene expression.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
The promoter region of the human factor IX gene showing the androgen response element (ARE) and the regions implicated in binding LF-A1/HNF4, C/EBP. Coordinates are relative to the transcription start site (+1). Haemophilia B Leyden mutations (closed arrows) lie outside the ARE but within the LF-A1/HNF4 and C/EBP regions implicated in constitutive expression. In contrast, the promoter mutation G-26C (haemophilia B Brandenburg, open arrow) causes lifelong haemophilia B and lies within both the ARE and the −9 to −40 region associated with constitutive expression.
“Certain mutations within the promoter of the factor IX gene disrupt or interfere with protein–DNA interactions in the promoter and so compromise the efficiency of transcription”
Before puberty, factor IX gene expression is regulated by constitutive promoter elements (C/EBP, LF-A1/HNF4, and possibly others). Mutations within these elements disrupt binding of the relevant proteins for transcription and haemophilia B results. At puberty, and thereafter, it is assumed that testosterone dependent transcription, mediated by the ARE, comes into play. Thus, provided that a promoter mutation does not lie within the ARE, amelioration of the haemophilia B is possible after puberty.
In contrast, promoter mutations that cause life long haemophilia B, such as haemophilia B Brandenburg136 (G-26C), have been found to lie within the region of overlap between the ARE and the LF-A1/HNF4 element and appear to interfere with both constitutive and hormonally regulated factor IX gene expression (fig 12).
Haemophilia A arising from mutations within the promoter of the factor VIII gene has not yet been demonstrated.
MUTATION DETECTION STRATEGIES
Studies aimed at defining the causative mutation within haemophilia cohorts have repeatedly shown that most patients possess a defect within the essential regions of the factor VIII or factor IX gene. These regions comprise the 5` promoter sequence, the exons, the intron–exon boundaries, and the 3` region around the polyadenylation signal sequence.
Therefore, the most direct strategy for mutation detection would be to amplify these regions from genomic DNA using PCR and screen the PCR products for a mutation. In the case of the factor IX gene, this is a straightforward task: the gene has only eight exons, the largest of which is less than 2 kb, so that fewer than 10 amplifications are required to cover the essential regions of the gene.105
In contrast, the large size and complexity of the factor VIII gene would necessitate almost 30 amplifications of genomic DNA to cover the essential regions. This is manageable and is the strategy used in some laboratories.75,140–142 Alternatively, factor VIII mRNA can be used to screen exons 1 to 13 and 15 to 26.143 The large exon 14 and the 5` promoter and 3` polyadenylation signal region can be screened from genomic DNA.143 Factor VIII mRNA is not obtained from the accepted primary site of transcription (the liver), but from circulating white blood cells in which the transcript is believed to be present as a result of ectopic (leaky) expression.143
Various mutation detection techniques can be used to screen the PCR products from the factor VIII or factor IX genes.75 One of the most informative is chemical mismatch cleavage,144 the approach used for the characterisation of haemophilia B mutations within the Swedish population145 and within the UK population.146 It has also been used extensively in the current effort to characterise the mutations within the UK haemophilia A population.147 Solid phase, fluorescence based chemical mismatch cleavage has provided a rapid route to screening the essential regions of the complex factor VIII gene in large numbers of patients.148
Direct nucleotide sequence analysis using automatic DNA sequencers is becoming more mainstream142 and confident results can be expected for male DNA (hemizygous) samples. Sequencing should be interpreted cautiously for female DNA because heterozygosity may fail to show in the sequencing data. Multiplex amplification of all of the essential regions of the factor IX gene in a single PCR, followed by sequencing,149 represents a further step forward and one that could be applied to the factor VIII gene.
MUTATIONS AND THE RISK OF INHIBITORS
The production of neutralising antibodies in response to infused factor VIII or factor IX has always been of considerable interest, principally because it is a major complication of replacement treatment.150,151 It is also potentially a major complication of gene therapy; therefore, insight into the factors that predispose towards inhibitor production would be of considerable value. Evidently, the mutation underlying the haemophilia is important141,150–153: mutations associated with the absence of a gene product, such as deletions or nonsense mutations, confer a high risk for inhibitor production; mutations associated with the presence of a gene product (even very low amounts of the protein) confer a low risk for inhibitor production. However, in reality, the situation is not so clear cut. Among patients with identical mutations, some may produce inhibitors and others may not.141,154 Clearly other factors are implicated. The possibility that these may include the genotype at the (MHC) locus has been investigated in patients with severe haemophilia A, with and without the intron 22 inversion.155,156 Only a weak association between human MHC (HLA) class II genotype and the development of inhibitor antibodies against factor VIII was obtained; this was slightly more pronounced in patients with the inversion than in those without.
Subtle effects may attenuate the relation between a haemophilia mutation and the risk of inhibitor production. For example, one would expect small insertions or deletions resulting in an alteration of the protein translation reading frame (frameshift mutations) to confer a high risk of inhibitor production. However, several groups have found the absence of inhibitors among patients with haemophilia A possessing a small deletion or insertion within a run of the same nucleotide.141,157 A proposed explanation is that transcriptional or translational errors may lead, fortuitously, to the occasional correction of the reading frame and this may lead to sufficient endogenous factor VIII to explain immune tolerance.141,157,158 Patients with a small deletion or insertion not within a run of the same nucleotide may not be so lucky.141
It therefore appears, at present, a difficult task, even with the knowledge of a patient's causative mutation, to predict an inhibitor response. Fortunately, however, inhibitors are a rare phenomenon among patients with haemophilia (apparently rarer in haemophilia B than haemophilia A). As one might expect, they are principally found within the severe group (high probability of the absence of a gene product), where they are found in 10–15% of individuals.150,153
FACTOR VIII AND FACTOR IX TURNOVER
The steady state concentrations of factor VIII and factor IX in the circulation reflect a balance of synthesis and removal. The clearance mechanism for both factors appears to be mediated, at least in part, by the low density lipoprotein receptor related protein (LRP), a liver multiligand endocytic receptor. Although the interaction of factor VIII with LRP is not disputed, there is some controversy concerning the region of factor VIII involved in binding and the effect of von Willebrand factor on the interaction. Factor VIII may bind LRP via a region within the light chain159 and this may be inhibited by von Willebrand factor,159 which also binds factor VIII via the light chain.118 This could then explain the beneficial effect of von Willebrand factor on the in vivo survival of factor VIII.16,17 Alternatively, factor VIII may bind LRP via the A2 domain in an interaction that is not influenced by von Willebrand factor.160 In vivo studies in von Willebrand factor deficient mice suggest that the accelerated clearance of factor VIII seen in the absence of von Willebrand factor may be the result of LRP mediated factor VIII catabolism.161 Clearly, LRP mediated removal of factor VIII appears to be real, and whether or not this is modulated by von Willebrand factor remains to be clarified.
LRP may also be involved in the catabolism of factor IX. Factor IX interaction with LRP appears to require activation of the coagulation factor by activated factor XI.162 Delivery of activated factor IX to LRP appears to require proteoglycans on the cell surface.162 In vitro, degradation of factor IX by cells deficient in either LRP or proteoglycans was significantly decreased compared with cells expressing these proteins. The relevance of these findings has yet to be established in vivo, although they suggest that one route to catabolism of activated factor IX may be via a proteoglycan on the cell surface, which delivers factor IX to LRP, which then targets the factor IX to the intracellular degradation pathway.
“Insights gained from mutations and their effects—the molecular pathology of disease—have revealed both subtle and major complexities at the genetic and protein levels”
CONCLUDING COMMENTS
The exploration of the factor VIII and factor IX genes and their gene products over the past 15 years has provided enormous amounts of information and has expanded into many and varied territories. To review all of these comprehensively would require several volumes; therefore, this review has necessarily been restricted. The areas covered reveal the ingenuity of the research community (scientist and clinician alike) and humankind's incessant struggle for knowledge and understanding. As each question is answered, and new questions arise, the resolution of our understanding sharpens.
Insights gained from mutations and their effects—the molecular pathology of disease—have revealed both subtle and major complexities at the genetic and protein levels. Similarly, insights gained from polymorphisms and their ethnic variations hint at a complex evolution awaiting further exploration. Finally, insights gained from the investigation of patients' genetics and the aetiology of the inhibitor complication are paving the path for gene therapy in previously untreated patients. The molecular biology of the haemophilias has provided food for research and food for thought, and the feast seems certain to continue into the distant future.
APPENDIX A DEFINITIONS OF POLYMORPHISMS FOUND IN THE FACTOR VIII AND FACTOR IX GENES, AND PROPERTIES RELEVANT TO LINKAGE STUDIES IN THE HAEMOPHILIAS
Single nucleotide polymorphisms
Single nucleotide polymorphisms (SNPs) are precisely what their name suggests: nucleotide positions at which different nucleotides are present in the gene in the normal population. Typically, they are biallelic (one of two nucleotides is present within any given gene).
Many SNPs fortuitously reside within recognition sequences for restriction endonucleases and this provides a convenient means by which to genotype them. Such SNPs are better known as restriction fragment length polymorphisms (RFLPs).
SNPs can be intronic or exonic. Those within exons can either be silent in that they do not change the amino acid encoded by the codon in which they occur, or they can change a codon so that a different amino acid is specified. The latter give rise to amino acid polymorphisms within the protein sequence.
Microsatellites
Microsatellites are sequences built up from multiple repeats of a given sequence motif and are polymorphic in that the number of repeats present within the gene varies in the normal population. They are typically multiallelic. The human genome contains a diversity of microsatellites, just one example of which is the sequence (CA)n, where “n” represents a variable number of repeats among different genes in the normal population.
Frequency
The alleles of SNPs and microsatellites occur with specific frequencies in the general population. In the case of biallelic SNPs, the equal occurrence of both alleles would result in a relative frequency of 0.5 for each. However, in reality, such equivalence in frequency between both alleles is rarely seen and a preponderance of one allele over the other is more usual—for example, (0.1/0.9) or (0.4/0.6) (allele 1/allele 2).
In the case of microsatellites, several alleles are usually present within the general population and their relative frequencies are not equal. One, two, or three alleles may predominate and other alleles may be more rare. The existence of several alleles is an important property of microsatellites in the context of linkage analysis (see later).
The allele frequencies of any polymorphism may vary between people from different racial backgrounds and, indeed, some loci may be polymorphic within one racial group but non-polymorphic within another.
Heterozygosity, homozygosity, and hemizygosity
Females with a normal karyotype (46:XX) have two copies of each polymorphism located on the X chromosome and this, of course, includes those polymorphic loci found within the factor VIII and factor IX genes. For a given polymorphic locus within these genes, a female may have identical alleles (homozygous) or she may have one allelic variant in one gene and a different allelic variant in the other (heterozygous).
The probability of heterozygosity can be estimated from the allele frequencies in the general population. For example, a biallelic locus for which the alleles occur at frequencies of 0.1/0.9 can be expected to show the following distribution of genotypes:
Homozygous (allele 1/allele 1): 0.1 × 0.1 = 0.01.
Heterozygous (allele 1/allele 2): 0.1 × 0.9 = 0.09.
Heterozygous (allele 2/allele 1): 0.1 × 0.9 = 0.09.
Homozygous (allele 2/allele 2): 0.9 × 0.9 = 0.81.
Thus, the expected frequency of heterozygosity (the rate of heterozygosity) is 0.09 + 0.09 = 0.18. Another way of interpreting this is that 18% of females may be expected to be heterozygous at that locus. An identical calculation done using a biallelic locus with allele frequencies of 0.5/0.5 yields a rate of heterozygosity of 0.5. This represents the maximum possible rate of heterozygosity for a biallelic locus and it can be interpreted to mean that 50% of females can be expected to be heterozygous at that locus.
Microsatellites yield much higher rates of heterozygosity because there are greater numbers of allelic variants and co-inheritance of identical alleles is intrinsically less probable than for a biallelic locus. For example, in a microsatellite with three allelic variants showing frequencies of 0.1 (allele 1), 0.6 (allele 2), and 0.3 (allele 3), the expected distribution of genotypes is as follows:
Homozygous (allele 1/allele 1): 0.1 × 0.1 = 0.01.
Homozygous (allele 2/allele 2): 0.6 × 0.6 = 0.36.
Homozygous (allele 3/allele 3): 0.3 × 0.3 = 0.09.
Heterozygous (allele 1/allele 2): 0.1 × 0.6 = 0.06.
Heterozygous (allele 2/allele 1): 0.6 × 0.1 = 0.06.
Heterozygous (allele 1/allele 3): 0.1 × 0.3 = 0.03.
Heterozygous (allele 3/allele 1): 0.3 × 0.1 = 0.03.
Heterozygous (allele 2/allele 3): 0.6 × 0.3 = 0.18.
Heterozygous (allele 3/allele 2): 0.3× 0.6 = 0.18.
From these calculations, the expected rate of heterozygosity is 0.06 + 0.06 + 0.03 + 0.03 + 0.18 + 0.18 = 0.54. This exceeds the 0.5 maximum for biallelic loci and yet it has only involved a locus with three alleles, one of which is of very low frequency (allele 1, 0.1). In practice, microsatellites have many more than three alleles and it is not unusual to observe rates of heterozygosity approaching 0.8.
A male with a normal karyotype (46:XY) only has one factor VIII gene and factor IX gene, therefore he will have only one copy of a given polymorphic locus and only one allele will be present (hemizygous).
Informativity and non-informativity
The outcome of a linkage study cannot be predicted at the outset: the data must be generated and then interpreted to reveal whether the polymorphic loci investigated have permitted a defective (or normal) gene to be tracked through the family. Any polymorphic loci that permit gene tracking from one individual to another within a family are said to be informative for that individual. Conversely, those loci that do not permit the gene to be tracked from one individual to another are non-informative for that individual.
Evidently, heterozygosity is associated with informativity: the two genes can be discerned by virtue of their different polymorphic alleles and this permits them to be tracked separately. Homozygosity is associated with non-informativity because the two genes cannot be distinguished. However, homozygosity can be helpful in establishing haplotypes from genotypes (see below).
Genotypes and haplotypes
Analysis of polymorphic loci to determine the alleles present generates a genotype for each individual in a family. This must then be resolved into haplotypes (the alleles in one gene separated from those in the other). In the case of the haemophilias, this is made easier by virtue of hemizygosity in males: the genotype is the haplotype in the case of hemizygosity. This may then permit haplotypes in females to be established by subtraction of a known haplotype from the genotype to give the second haplotype.
Homozygosity in females also permits the determination of haplotypes: if a female is homozygous, both genes must have identical haplotypes for the polymorphic loci examined, and this may permit further haplotypes to be resolved within the family.
In phase
The haplotype found in the factor VIII or factor IX gene of the male haemophiliac within a family is said to be in phase with haemophilia: it identifies the defective factor VIII or factor IX gene. It is worth stressing that this haplotype undoubtedly exists in the normal population where it is not associated with a defective gene, it just so happens that, in the family being investigated, the defective gene has this haplotype. Thus, discovery of the same haplotype in other members of the family does not necessarily define the presence of the defective gene. Inheritance of the defective gene has to be shown directly by analysis of relevant family members.
Linkage disequilibrium
Polymorphisms within a given gene are necessarily linked. When a certain allele at one locus always occurs in conjunction with a certain allele at another locus, the loci are said to show complete linkage disequilibrium. When the alleles at one locus occur entirely independently of the alleles at a second locus, the loci are said to show complete linkage equilibrium. Between these two extremes there is a continuous scale of linkage disequilibrium.
If two loci show complete linkage disequilibrium, the information obtained at one will be identical to that obtained at the other and no additional information will be gained by examination of both. If one locus is homozygous and non-informative, the other one will be also, so that informativity is not increased by investigation of both loci. For this reason it may seem redundant to investigate loci showing linkage disequilibrium. However, examination of both does permit corroboration of the information obtained from either. This can be valuable, lending confidence to the data generated in the study. Loci that are in linkage disequilibrium may therefore not increase the informativity of a study very much, but they do provide a double check of the data, and this may be invaluable.
Implications of these properties of polymorphisms
The goal of a linkage study in the haemophilias is to establish carrier status and/or offer prenatal diagnosis with confidence. This requires an informative genetic result, and this should be derived preferably from more than one informative polymorphic locus. For this to be achieved, the best loci to examine are those with the highest rates of heterozygosity and the lowest degree of linkage disequilibrium.
APPENDIX B DATABASE ENTRIES AT THE NATIONAL CENTRE FOR BIOTECHNOLOGY INFORMATION (URL HTTP://WWW.NCBI.NLM.NIH.GOV/)
Factor VIII (F8C)
-
Gene: NT_025284
-
mRNA: NM_000132
-
Protein: NP_000123
-
Locus Link: LocusID 2157
F8A
-
Gene: NT_025307
-
mRNA: NM_012151
-
Protein: NP_036283
-
Locus Link: LocusID 8263
F8B
-
Gene: NT_025284
-
mRNA: M90707
-
Protein: AAA58466
-
Locus Link: LocusID 2157
F8 Int22h-1
-
DNA: X86011 and X86012
Factor IX (F9)
-
Gene: K02402
-
mRNA: NM_000133
-
Protein: NP_000124
-
Locus Link: LocusID 2158
REFERENCES
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵