Article Text

Abstract
CpG islands are GC rich sequences that are found in the promoters of many genes in higher eukaryotes. They contain a high frequency of CG dinucleotides, which are substrates for DNA methylases. Methylation leads to transcriptional silencing of promoters. Owing to their high GC content CpG islands exhibit strong base–base interactions, which lead to superstructures and consequently to regions with higher melting temperatures. Therefore, Taq polymerases (especially sequenases) fall off their templates, causing premature termination of the polymerase chain reaction (PCR) or sequencing reactions. The results from such reactions are thus insufficient for further analysis. Therefore, we have evaluated the use of 7-deaza-2`-deoxyguanosine for PCR amplification of the human p16INK4A promoter and sequencing of HUMARA exon 1 PCR products. Our results show that the addition of 7-deaza-2`-deoxyguanosine significantly improves results, particularly when small amounts of poor quality DNA are available as starting material.
- BD, Big Dye
- deaza-dGTP
- 7-deaza-2`-deoxyguanosine
- MSP, methylation specific PCR
- PCR, polymerase chain reaction
Statistics from Altmetric.com
- BD, Big Dye
- deaza-dGTP
- 7-deaza-2`-deoxyguanosine
- MSP, methylation specific PCR
- PCR, polymerase chain reaction
CpG islands are GC rich sequences, which are found in more than 60% of gene promoters in higher eukaryotes. They are target structures for DNA methylases—cytosines located 5` of guanosine are the only bases methylated in higher eukaryotes.1 Methylation of CpG islands occurs during imprinting or inactivation of X chromosomes,2 and as aberrant methylation of normally unmethylated CpG islands in transformed and immortalised cells.3 One approach to detect the methylation of CpG islands is a combination of restriction enzyme digestion using methylation sensitive enzymes and subsequent polymerase chain reaction (PCR). This type of assay can be used to determine clonality by analysing exon 1 of the HUMARA gene, which encodes the human androgen receptor.2 However, major problems arising with this type of assay are incomplete restriction and low sensitivity. To overcome this problem, methylation specific PCR (MSP) was developed; this technique uses a combination of chemical DNA modification with bisulphite and subsequent PCR. Bisulphite treatment leads to deamination of unmethylated cytosine residues to yield uracil, so that changes in the DNA sequence are generated. Thus, by defining appropriate primer sets methylated and unmethylated DNA can be discriminated by PCR if bisulphite treated templates are used. Because the primer pairs are specific for methylated or unmethylated DNA, respectively, MSP is a very sensitive technique. MSP has been described for the determination of the methylation status of the human p16INK4A promoter, which encodes an inhibitory protein of the cell cycle with a molecular mass of 16 kDa (inhibitor of kinase 4).4 Because CpG islands are GC rich they are prone to form superstructures and display higher melting temperatures. Thus, it is difficult to generate PCR products or to obtain a readable sequence from such PCR products.5 Here, we show that the use of 7-deaza-2`-deoxyguanosine (deaza-dGTP) in PCR reactions allows the generation of full length PCR products of the human p16INK4A promoter. Furthermore, readable sequences from PCR products of the HUMARA exon 1 were obtained. The addition of deaza-dGTP was particularly helpful when working with low amounts of DNA template of poor quality.
“Methylation of CpG islands occurs during imprinting or inactivation of X chromosomes”
MATERIAL AND METHODS
Purification of DNA templates
DNA from cultured SW1116 (human colorectal adenocarcinoma cell line; number 87071006; ECACC, London, UK) or ARH77 (human B cell lymphoma line; CRL1621; ATCC, Rockville, Maryland, USA) cells was isolated according to the instruction manual of the QIAamp DNA mini kit (Qiagen, Hilden, Germany). Both cell lines were cultured according to standard procedures in DMEM (SW1116) or RPMI 1640 (ARH77) containing 50μM 2-mercaptoethanol and 10% (vol/vol) fetal calf serum (all Life Technologies, Karlsruhe, Germany). After purification DNA concentration and quality were determined by UV photometry.
PCR conditions
We amplified the human p16INK4A promoter (accession number X94154) and HUMARA exon 1 (accession number M27423) according to previously published methods.2,4 The PCR mixtures contained 1× PCR buffer (Taq polymerase: 50mM KCl, 100mM Tris/HCl, pH 9.0, 0.1% (vol/vol) Triton-X 100, or AmpliTaq Gold polymerase: PCR Gold buffer (Applied Biosystems, Weiterstadt, Germany)), 1.5mM (p16INK4A) or 2.0mM (HUMARA) magnesium chloride, 200μM dNTP mix (both) or deaza-mix (deaza-dGTP instead of dGTP), 10 pmol of each primer (p16INK4A WT4, HUMARA2), 1 U Taq polymerase (Promega, Heidelberg, Germany) or for hot start PCR AmpliTaq Gold (Applied Biosystems) and 50 ng template (p16INK4A, SW1116; HUMARA, ARH77) in a final volume of 25 μl. The following PCR cycle profiles were used.
-
p16INK4A: five minutes (Taq polymerase) or 12 minutes (AmpliTaq Gold) at 94°C; 35 cycles of 20 seconds at 94°C, 20 seconds at 65°C, and 20 seconds at 72°C; followed by two minutes at 72°C.
-
HUMARA: five minutes (Taq polymerase) or 12 minutes (AmpliTaq Gold) at 94°C; 35 cycles of 20 seconds at 94°C, 20 seconds at 63.6°C, one minute at 65°C; followed by six minutes at 72°C on a Hybaid MBS 0.25 PCR machine (Hybaid, Heidelberg, Germany). The PCR products were analysed on 3% (wt/vol) 1× TBE agarose gels.
Sequencing conditions
For sequencing of HUMARA exon 1 PCR products residual primers and nucleotides were removed using ammonium acetate precipitation (one volume of PCR reaction, one volume of 4M ammonium acetate (Sigma, München, Germany), and six volumes of 100% (vol/vol) EtOH; 15 minutes of maximum speed in a microcentrifuge). The precipitate was redissolved in 20 μl water and used as a template for further sequencing using the Big Dye (BD) sequencing kit (Applied Biosystems) (4 μl BD premix, 2 μl purified PCR product, 10 pmol primer (see above)) in a final volume of 20 μl using the following cycle profile: 15 cycles of 10 seconds at 96°C and 90 seconds at 60°C on a Hybaid MBS 0.25 PCR machine (Hybaid). Excessive dye terminators were removed applying the Dye-Ex kit (Qiagen), according to the user's instructions. Finally, 2 μl of the eluate was mixed with 18 Hi-Di formamide (Applied Biosystems), denatured for two minutes at 90°C, and loaded on to a genetic analyser ABI310 (Applied Biosystems). Separation was done using a short capillary (30 cm) and POP6 (6% performance optimised polymer; Applied Biosystems). The resulting electropherograms were analysed on a personal computer using the freely available computer software CHROMAS (http://www.technelysium.com.au/chromas.html).
RESULTS AND DISCUSSION
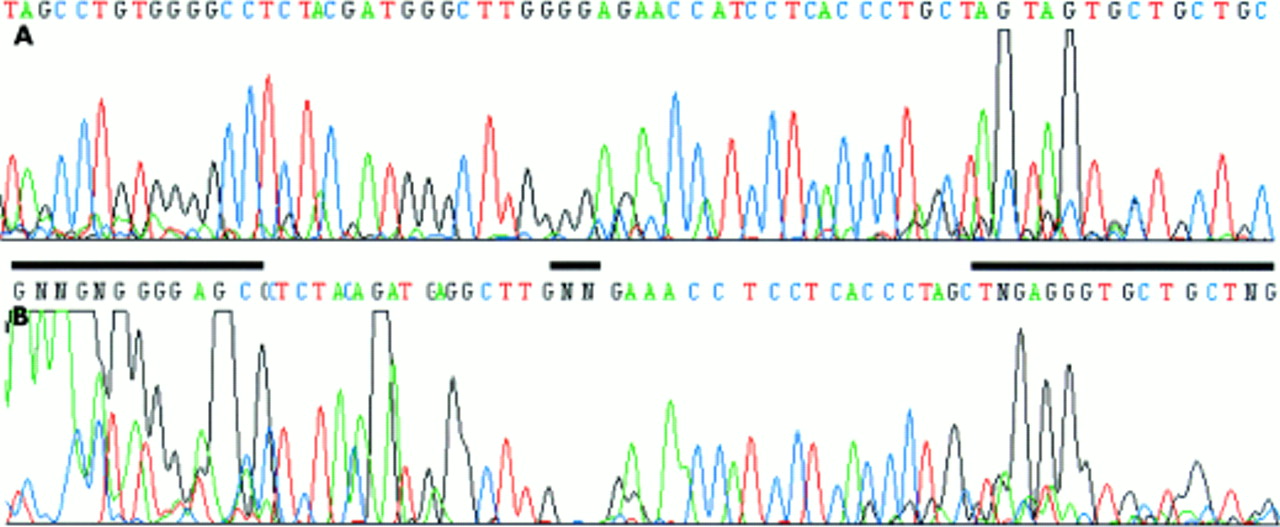
In the course of studies on the methylation status of the p16INK4A promoter from microdissected areas of colorectal adenocarcinomas we found it difficult to obtain PCR products using the published p16INK4A WT primers and reaction conditions.4 In separate experiments, we developed a clonality assay based on the HUMARA exon 1 locus, because the originally described method2 was too insensitive to analyse DNA from microdissected formaldehyde fixed tissue. However, both the PCR of p16INK4A and sequencing of PCR products from exon 1 of the HUMARA locus did not yield reproducible results. The 140 bp PCR product of the p16INK4A promoter was generally faint or invisible (fig 1B). Similarly, large stretches of the HUMARA exon 1 sequence were not readable (fig 2B). Variations of the standard parameters—magnesium concentration, melting temperature, denaturing temperature, hot start using AmpliTaq Gold—did not solve these problems. Because of the high GC content of these sequences (p16INK4A, 78%; HUMARA, 65%) we tried several additives, such as DMSO,6 formamide,7 and betaine,8 which had been reported to improve PCRs of GC rich structures. However, these did not improve the results. 7-Deaza nucleotide analogues have been reported to weaken base–base interactions, thus solving superstructures and improving PCRs.5 Therefore, we added deaza-dGTP instead of dGTP to the PCRs. Independently of hot start, a PCR product could be generated for the p16INK4A promoter (fig 1A). Improvement was also seen for sequencing of deaza-dGTP containing PCR products of the HUMARA exon 1 (fig 2B). All experiments were carried out at least in triplicate and the addition of deaza-dGTP is now a routine procedure in our laboratory. Thus, deaza-dGTP is very useful when performing PCR and subsequent sequencing of GC rich sequences if low amounts of template are used and/or DNA quality is poor, particularly from microdissected human tissue.
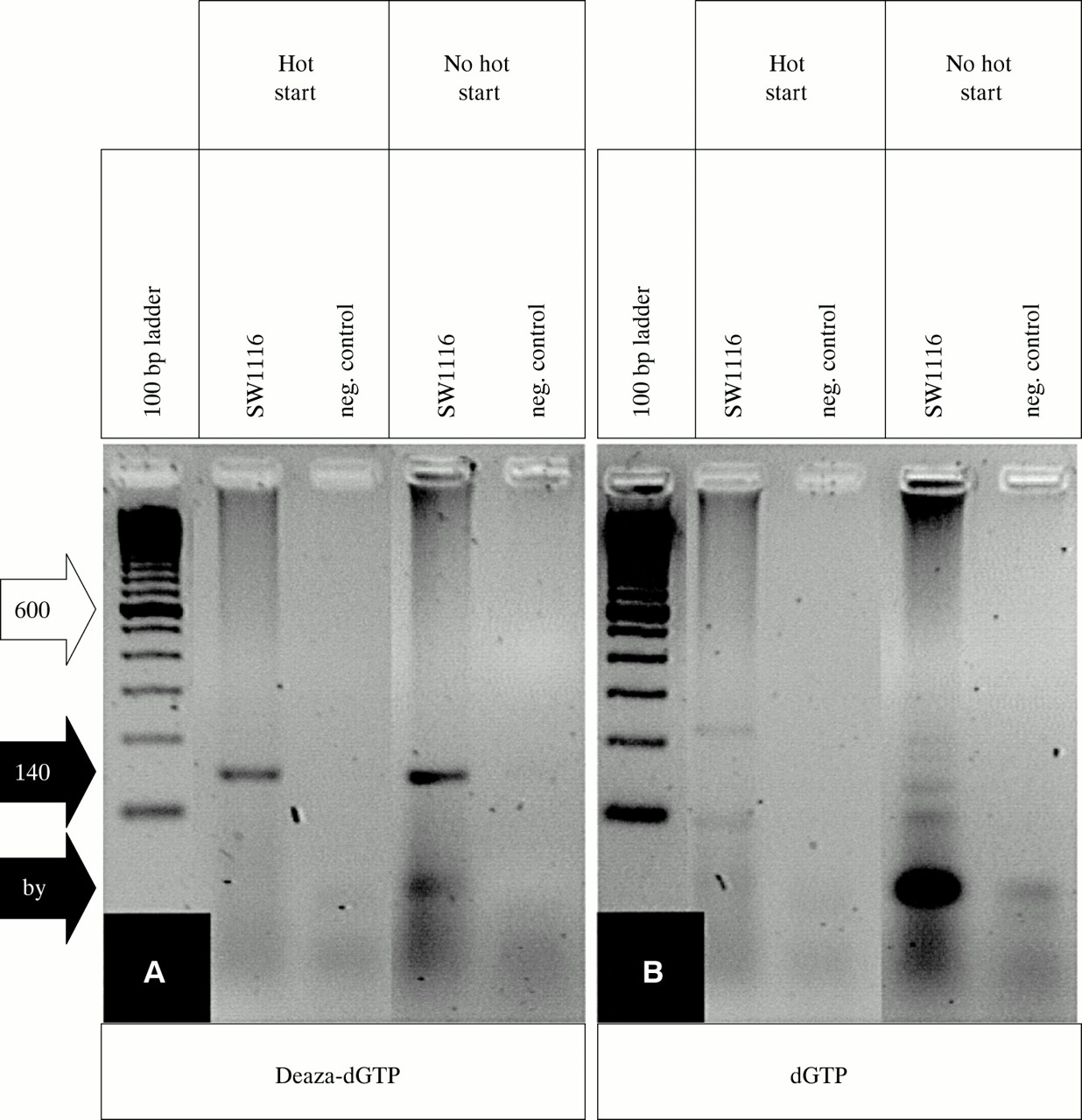
(A) 7-Deaza-2`-deoxyguanosine (deaza-dGTP) allows PCR of the human p16INK4A promoter because the specific 140 bp product (black arrow, 140) is clearly visible whether standard Taq polymerase (no hot start) or AmpliTaq Gold (hot start) is used. (B) dGTP gives rise to unspecific byproducts (black arrow, by), which are reduced if hot start PCR is performed. The 600 bp band of the 100 bp ladder standard is indicated (white arrow, 600).

{kind=link}
{kind=link}
(A) The presence of 7-deaza-2`-deoxyguanosine in PCR products of the HUMARA exon 1 allows subsequent sequence reactions. (B) The same sequence as in (A) except that dGTP was used for generating PCR products. GC rich regions that are unreadable are indicated by the black bars.
Take home messages
-
Because of the high GC content of CpG islands it is difficult to generate polymerase chain reaction (PCR) products from these regions or to obtain a readable sequence from such PCR products
-
The addition of 7-deaza-2`-deoxyguanosine significantly improves the PCR amplification of the human p16INK4A promoter and sequencing of HUMARA exon 1 PCR products, both of which have a high GC content
-
The addition of 7-deaza-2`-deoxyguanosine is particularly useful when small amounts of poor quality DNA are available as starting material
“7-Deaza nucleotide analogues have been reported to weaken base–base interactions, thus solving superstructures and improving polymerase chain reactions”
Sequencing of GC rich sequences often leads to migration anomalies, especially band compressions. Therefore, many manufacturers add deaza analogues to their sequencing reaction mixes and in situations where commercial kits are not available the addition of deaza-dGTP and deaza-dITP is of great benefit.9 If DNA is isolated from formaldehyde fixed paraffin wax embedded tissue sections using microdissection poor quality and low amounts of DNA become an additional issue. The use of deaza-dGTP has been reported to improve PCR product yield,10 and this is confirmed by our study for the generation of PCR products from the p16INK4A promoter. In cases where additional sequencing is required it can be advantageous to use deaza-dGTP containing PCR products as templates, as we have shown for the HUMARA exon 1. Even without deaza-dGTP, very strong signals can be seen at the beginning of the sequence (fig 2B), but intensity drops towards the end of the sequence (fig 2B), obviously because of the premature termination of the sequenase reaction. This situation changes when deaza-dGTP is present in the PCR product (fig 2A). Thus, particularly when dealing with DNA extracted from microdissected tissue, the addition of deaza-dGTP is strongly recommended for the analysis of CpG islands.
Acknowledgments
We thank A Haynl and C Koch for expert technical assistance and G Niedobitek for critical reading of the manuscript and discussion. This work was supported by grants from Marohn Stiftung (Nie-Ma, Mikrodissektion) to AJ; Wilhelm-Sander-Stiftung (99.065.1) to AJ, TB, and TK; and BMBF-Pädiatrische Onkologie und Hämatologie- (DLR 01GI9962) to AJ.