


Abstract
The Rho GTPases are involved in actin cytoskeleton organization and signal transduction. They need polyisoprenylation for membrane association and activation. Lovastatin, a hydroxymethylglutaryl coenzyme A inhibitor, prevents isoprene synthesis and thereby lipid modification of the Rho protein carboxy terminus. Because lovastatin causes rounding up of cultured cells, we investigated whether the compound acts on the actin cytoskeleton through Rho proteins. Lovastatin treatment decreased F-actin content in a time- and concentration-dependent manner. G-actin content remained unchanged. In lovastatin-treated NIH 3T3 cells, the amount of Rho protein which was ADP-ribosylated by Clostridium botulinum exoenzyme C3 decreased in membranes and increased in the cytosol fraction. Cycloheximide prevented lovastatin-induced rounding up of cells. However, after microinjection or direct application of exoenzyme C3, cells treated with cycloheximide and lovastatin rounded up again. On the contrary, lovastatin-treated, round Swiss 3T3 cells reverted to a flat morphology when microinjected with dominant active RhoA (Val14RhoA). Escherichia coli cytotoxic necrotizing factor (CNF1) which activates Rho proteins caused flattening of round, lovastatin-treated NIH 3T3 cells. These results suggest that lovastatin affects the actin cytoskeleton through inactivation of Rho proteins.
Rho-family proteins (RhoA, B, C, Rac1, 2 and Cdc42) participate in regulation of the actin cytoskeleton (Paterson et al., 1990; Chardinet al., 1989) (for review see Machesky and Hall, 1996). They appear to maintain cell shape by organizing the actin cytoskeleton and they change cell shape by regulating formation of membrane ruffles, lamellipodia and filopodia (Kozma et al., 1995; Nobes and Hall, 1995).
Recently, a possible signaling pathway between Rho proteins and the cytoskeleton was elucidated. After the interaction between activated RhoA and p164 Rho-kinase was demonstrated (Matsui et al., 1996), Rho-kinase was shown to phosphorylate and inactivate the myosin-binding subunit of myosin-phosphatase (Kimura et al., 1996). Thereby myosin light chain, its substrate protein and regulatory unit of the myosin complex, remains phosphorylated. The active myosin complex on its part can bind to actin. This results in cell contraction and possibly in stress fiber formation and cell adhesion (Chrzanowska-Wodnicka and Burridge, 1996). By a second mechanism Rho-kinase directly phosphorylates myosin light chain and thereby affects actomyosin interaction (Amano et al., 1996). Further, it has been suggested that Rho-dependent regulation of phospholipids takes part in actin cytoskeleton organization. Rho regulates phosphatidylinositol 4-phosphate 5-kinase and the phospholipids reportedly control numerous actin-binding proteins (Chonget al., 1994).
Rho proteins are involved in various signaling pathways. Rho proteins interact with protein kinases like protein kinase N (Watanabe et al., 1996) and p65 PAK kinase (Manser et al., 1994). Evidence has shown that they influence phospholipid turnover through phosphoinositide 3-kinase (Zhang et al., 1993), phosphatidylinositol 4-phosphate 5-kinase (Chong et al., 1994), phospholipase C-δ (Homma and Emori, 1995) and phospholipase D (Malcolm et al., 1994). Moreover, they affect transcription factor signaling pathways (Hill et al., 1995) and cell transformation (Khosravi-Far et al., 1996).
Various bacterial toxins modify Rho-family proteins in vivoand in vitro and have helped to gain insights into Rho protein functions. Clostridium botulinum exoenzyme C3 (Aktories et al., 1988) ADP-ribosylates and thereby inactivates RhoA, B and C (Aktories et al., 1992).Clostridium difficile toxins A and B have a broader spectrum and inactivate Rho, Rac and Cdc42 by glucosylation (Just et al., 1995). Both types of toxins lead to destruction of the actin cytoskeleton, and thereby cause cell rounding. On the other side, cultivated cells treated with Escherichia coli cytotoxic necrotizing factors, CNF1 or CNF2, form bundles of stress fibers (Oswald et al., 1994; Falzano et al., 1993). This has recently been found to be caused by deamidation of Gln63 of Rho proteins by CNF1 (Schmidt et al., 1997). Thereby Rho proteins are converted to dominant active Glu63Rho.
Rho-family proteins are posttranslationally isoprenylated at the carboxy-terminal “CAAX-box” (Goldstein and Brown, 1990). C represents cysteine, A an aliphatic amino acid and X any amino acid. RhoA and RhoC are geranylgeranylated and RhoB is geranylgeranylated or farnesylated by specific transferases (Adamson et al., 1992;Armstrong et al., 1995). After covalent binding of these polyisoprenoid residues, the three carboxy-terminal amino acids are cleaved and the carboxy terminus is carboxymethylated. The lipid modifications appear to be important for membrane binding of Rho-family proteins and for interaction with RhoGDI (Hori et al., 1991). The farnesyl and geranylgeranyl residues are generated during cholesterol biosynthesis (Goldstein and Brown, 1990). HMG-CoA reductase regulates this pathway and generates the precursor mevalonic acid from HMG-CoA.
Lovastatin, isolated from Aspergillus andMonascus species, is a competitive inhibitor of HMG-CoA reductase and is used as a therapeutic agent to treat hypercholesterolemia (Grundy, 1988). HMG-CoA reductase inhibitors also arrest cell replication (Quesney-Huneeus et al., 1979); they inhibit histamine release from rat peritoneal mast cells (Rocheet al., 1995), serotonin release from RBL-2H3 cells, a rat basophilic leukemia cell line (Deanin et al., 1991) and diminish insulin secretion from HIT-T15 cells (Li et al., 1993). They induce cell rounding which is independent of cellular cholesterol but is reversed by mevalonic acid (Schmidt et al., 1982). It is postulated that HMG-CoA reductase inhibitors cause cell rounding by prevention of isoprenylation of proteins important for cytoskeletal organization (Bifulco et al., 1993). Here we investigated whether lovastatin acts on the cytoskeletonvia the polyisoprenylated Rho-family proteins.
Materials and Methods
Materials.
[32P]NAD was obtained from DuPont-NEN (Dreieich, Germany). C. botulinum C2 toxin (Ohishi et al., 1980), C. botulinum C3 toxin (Aktories et al., 1988) and C. difficile toxin B (Just et al., 1997) were purified as described. E. coli CNF1 was prepared by sonication of E. coli cells carrying the cosmid vector pISS391 containing the CNF1 gene in buffer A (Tris-HCl, 50 mM, pH 7.4; phenylmethylsulfonyl fluoride, 1 mM; aprotinin, 0.4 mg/ml; leupeptin, 0.25 mg/ml; benzamidine, 0.8 mg/ml) and sterile filtration of the lysate (Falbo et al., 1993). Lovastatin was kindly donated by Merck, Sharp and Dohme (Munich, Germany) and was activated as described (Kita et al., 1980). Recombinant Val14RhoA was prepared as described (Just et al., 1994). All other chemicals were of analytical grade and were obtained from commercial sources.
Cell culture.
NIH 3T3 and Swiss 3T3 mouse fibroblasts were grown in Dulbecco’s modified Eagle’s medium supplemented with 10% fetal calf serum, 4 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin in a humidified atmosphere of 5% CO2 at 37°C. For the experiments, cells were grown to subconfluence in 3.5-cm Petri dishes. Sterile solutions in culture medium of the compounds and toxins tested were added for times and in concentrations given in the figure legends. New NIH 3T3 cells were cultivated from a frozen stock every 3 months to maintain cell lines with constant characteristics. Cells were observed by use of a Zeiss Axiovert microscope (Zeiss, Oberkochem, Germany) and photographed with Agfapan film.
Quantification of filamentous actin content in NIH 3T3 cell lysates.
Cellular F-actin content was determined as described (Suttorp et al., 1991). NIH 3T3 cells were grown as described above and treated with lovastatin for the times and with the concentrations given in the text and figure legends. After removal of culture medium, cells were rinsed twice with ice-cold buffer C (75 mM KCl, 3 mM MgSO4, 1 mM ethyleneglycol-bis(β-aminoethyl ether)-N,N,N′,N′-tetraacetic acid, 0.2 mM dithiothreitol, 10 μg/ml aprotinin, 0.1 mM phenylmethylsulfonyl fluoride and 10 mM imidazole, pH 7.2) permeabilized with saponin (0.03%) in buffer C for 10 min at room temperature, washed twice with buffer C and fixed with formaldehyde (3% in buffer C) for 20 min at room temperature. Again, cells were washed twice and stained with NBD-phalloidin (0.5 ml, 0.175 μg/ml in buffer C) for 30 min at room temperature. After washing of cells, NBD-phalloidin was eluted by incubation with ice-cold methanol (0.6 ml) for 6 h at −20°C. The amount of NBD-phalloidin eluted was measured with a fluorescence spectrometer at an excitation wavelength of 497 nm and an emission wavelength of 527 nm and was directly proportional to cellular F-actin content.
Quantification of monomeric actin in NIH 3T3 cell lysates.
Rabbit skeletal muscle α-actin was purified as described (Spudich and Watt, 1971). Monomeric actin (G-actin) in the calcium and ATP-bound form was purified by Sephacryl S-200 chromatography in buffer G (triethanolamine-HCl 10 mM, pH 7.5; CaCl2, 0.2 mM; ATP, 0.2 mM; NaN3, 3 mM). Actin concentrations were determined photometrically at 290 nm with an absorption coefficient of 24,900 M−1cm−1 (Wegner, 1976).
NIH 3T3 cells were grown to subconfluence in culture medium as described above. Thereafter, medium was changed and cells were incubated with C. botulinum C2 toxin, C. limosumC3-like exoenzyme (a fusion toxin for better uptake which will be described elsewhere), C. difficile toxin B and lovastatin for the times and with the concentrations given in the legend of figure2C. For determination of G-actin content, cell culture medium was removed, cells were placed on ice, rinsed twice with ice-cold phosphate-buffered saline and scraped off in 0.1 ml of buffer B (potassium phosphate, 5 mM, pH 7.6; NaCl, 150 mM; MgCl2, 2 mM; dithiothreitol, 0.1 mM; ATP, 0.2 mM; Triton X-100 0.5%; phenylmethylsulfonyl fluoride, 1 mM). An aliquot of the lysates was diluted 10-fold in buffer B and incubated for 10 min with an equal volume of a guanidine hydrochloride solution (guanidine hydrochloride, 1.5 M; sodium acetate, 1 M; CaCl2, 1 mM; ATP, 1 mM; Tris-HCl, pH 7.5, 20 mM). Lysates treated without and with guanidine hydrochloride were centrifuged (10 min, 4°C, 7000 × g) and the supernatant was used for further analysis. An aliquot was removed for protein content determination.
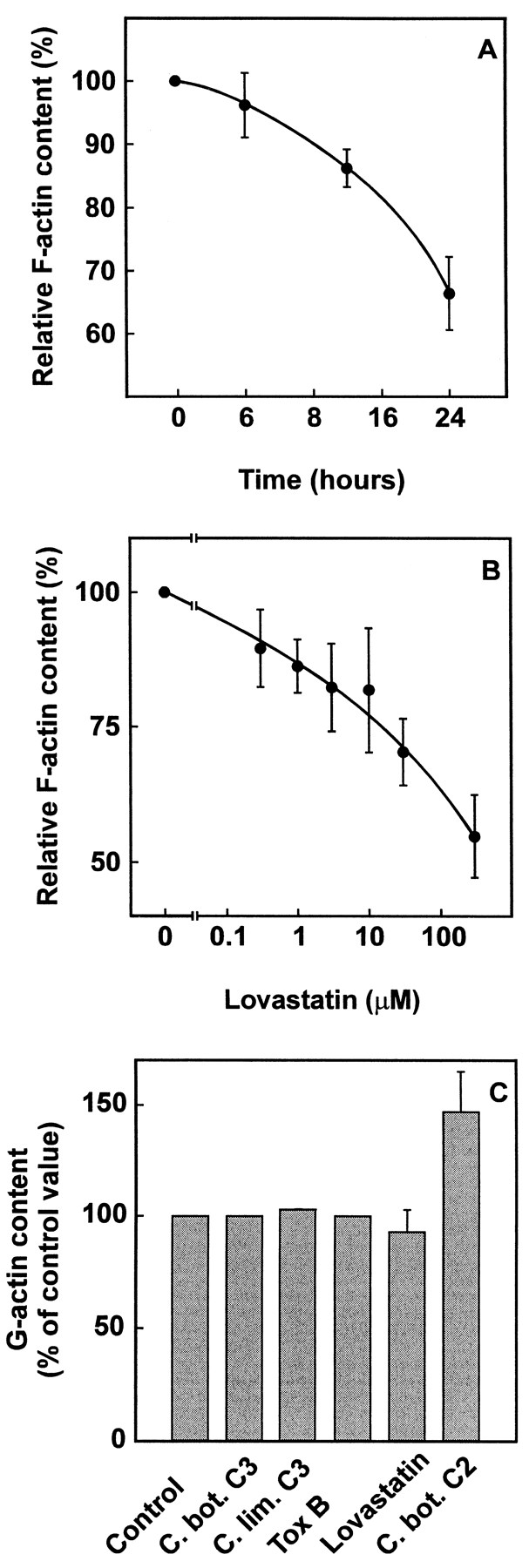
Effects of lovastatin and bacterial cytotoxins on F-actin and G-actin content of NIH 3T3 mouse fibroblasts. (A) NIH 3T3 cells were incubated with 30 μM lovastatin for 0, 6, 15 and 24 h. Cellular F-actin content was determined with NBD-phalloidin as described under “Materials and Methods.” F-actin content at the start of the experiment was set to 100% and is equivalent to 15.8 ± 0.86 absorption units/mg protein (mean ± S.E.M.). Data are mean values and S.E.M. from three independent experiments performed with duplicates. (B) NIH 3T3 cells were incubated with varying concentrations of lovastatin for 24 h and F-actin content was determined as described. F-actin content in the absence of lovastatin was set to 100% and was equivalent to 11.1 ± 0.41 absorption units/mg protein (mean ± S.E.M.). Data are mean values and S.E.M. from three independent experiments performed with duplicates. (C) NIH 3T3 cells were incubated without (Control) or with C. botulinum exoenzyme C3 (20 μg/ml, C. bot. C3), C. limosum C3-like exoenzyme (20 μg/ml, C. lim. C3) and lovastatin (30 μM) for 24 h; with C. difficiletoxin B (2 μg/ml, Tox B) for 90 min and with C. botulinum C2 toxin (C2 I 600 ng/ml, C2 II 1200 ng/ml, C. bot. C2) for 4 h. Cells were lysed and G-actin content was determined by DNase I inhibition assay as described under “Materials and Methods.” The relative G-actin content is given: G-actin content of control cells which were incubated with cell culture medium and vehicle only was set to 100%. Mean and S.E.M. values from three independent experiments performed with duplicates were calculated.
Cellular G-actin content was determined with the DNase I inhibition assay which is based on inhibition of DNase I enzymatic activity by G-actin (Blikstad et al., 1978). The reaction was performed in buffer D (Tris-HCl, 100 mM, pH 7.5; MgSO4, 4 mM; CaCl2, 1.8 mM) in a total volume of 1 ml containing DNA (40 μg/ml), DNase I from beef pancreas (1.5 μg/ml), cell lysate or buffer B, respectively, and buffer G or G-actin (0.7–1.5 μg/ml), respectively. Change of absorbance was measured continuously for 30 s at 260 nm. A standard curve was generated with rabbit muscle G-actin (about 0.7–1.5 μg) which resulted in 30 to 70% inhibition of DNase I activity. Concentrations of cell lysates were adjusted until inhibition of DNase I activity was also between 30 and 70%. Within this range, the relationship between G-actin content and inhibition of DNase I activity is linear.
Inhibition of DNase I activity was calculated according to the equation: [(Su −Si)/Su] × 100, where Si is the slope of the inhibited absorbance curve in the presence of varying G-actin concentrations or cell lysate and Su is the slope of the uninhibited absorbance curve in the absence of G-actin or cell lysate.
ADP-ribosylation assay.
Subconfluent NIH 3T3 cells in 3.5-cm dishes were treated with lovastatin, cycloheximide or CNF1 E. coli lysate for the times and with the concentrations given in the text or the figure legends. Thereafter, they were twice rinsed with ice-cold phosphate-buffered saline. Cells were scraped off in lysis-buffer (triethanolamine-HCl, 10 mM, pH 7.5; phenylmethylsulfonyl fluoride, 1 mM). Lysates were separated into cytosol and membrane fraction by centrifugation (60 min, 100,000 × g). Protein concentrations were adjusted after determination of protein content in an aliquot by Bradford analysis.
For ADP-ribosylation (Aktories et al., 1988) cytosol or membrane fraction were incubated with C. botulinum exoenzyme C3 (0.5 μg/ml) and [32P]NAD (0.1 μM, 0.8 μCi/tube) in ADP-ribosylation buffer (triethanolamine-HCl, 10 mM, pH 7.5; ethylenediaminetetraacetic acid, 1 mM; MgCl2, 2 mM; dithiothreitol, 1 mM;, phenylmethylsulfonyl fluoride, 1 mM) for 30 min at 30°C in a total volume of 50 μl. The reaction was terminated by addition of Laemmli sample buffer and proteins were separated by SDS-PAGE. Radioactivity incorporated was visualized by use of a phosphorimager (Molecular Dynamics, Sunnyvale, CA).
Microinjection studies.
Swiss 3T3 cells were seeded onto coverslips and grown to 70 to 80% confluence. Subsequently, they were treated with cycloheximide (5 μg/ml) in the absence or presence of lovastatin (30 μM) for 24 h. Thereafter, cells were microinjected with C. botulinum exoenzyme C3 (100 μg/ml) and photographs were taken after 1 h. In further experiments, Swiss 3T3 cells were preincubated with lovastatin (30 μM) for 24 h and microinjected with either buffer or Val14RhoA (1 mg/ml). Photographs of morphological changes were taken 1 h after microinjection. Microinjection was performed with an Eppendorf micromanipulator and microinjector (Eppendorf, Germany) with a Zeiss Axiovert microscope. The amount of sample injected was about 1% of total cell volume. Microinjection studies for each type of experiment were performed on at least six cells and on at least two independent occasions. Here, Swiss 3T3 instead of NIH 3T3 cells were used because effects of microinjected Val14RhoA on cell morphology were found to be exhibited better.
Determination of protein concentrations.
Protein content was quantified with the Bradford reagent with use of bovine serum albumin as standard (Bradford, 1976).
Results
Effects of lovastatin on morphology and subcellular Rho content of NIH 3T3 fibroblasts.
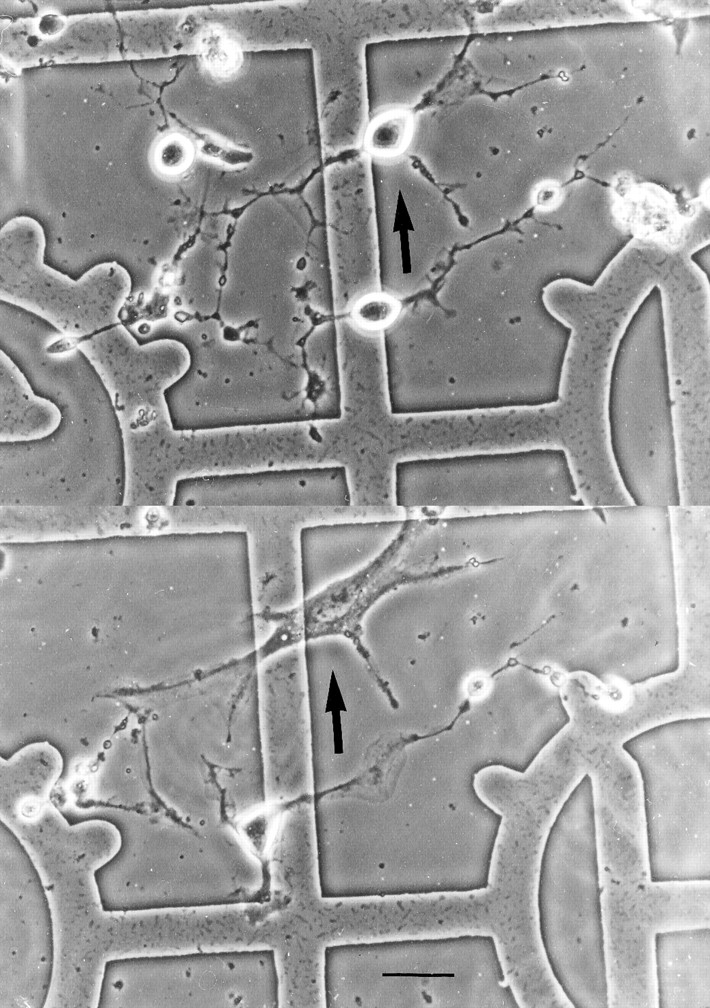
Compactin and lovastatin, both HMG-CoA reductase inhibitors, cause rounding up of Swiss 3T3 fibroblasts (Schmidt et al., 1982). Fig. 1shows that NIH 3T3 cells round up and form processes with varicosities in response to lovastatin (30 μM). Rounding up of cells was first observed about 6 h after addition of lovastatin (30 μM) and was complete after 15 h of treatment. Microscopy showed that lovastatin causes fragmentation of actin cables (Fenton et al., 1992); therefore, we studied the effects of lovastatin on F-actin and G-actin content by an NBD-phalloidin absorption and desorption technique. As shown in fig. 2A lovastatin decreased cellular F-actin content in a time-dependent manner. After 24 h of lovastatin (30 μM) treatment, cellular F-actin content was reduced to about 60% of the initial content. Also, lovastatin decreased F-actin in a concentration-dependent manner (fig.2B). A 50% inhibition of F-actin content was obtained with 300 μM lovastatin after 24 h of treatment.

Effects of lovastatin on morphology of NIH 3T3 mouse fibroblasts. NIH 3T3 mouse fibroblasts were incubated in the absence or presence of lovastatin (30 μM) for 24 h. Photomicrographs of untreated (upper panel) and treated (lower panel) cells are shown. Bar, 10 μm.
Concomitantly, cellular G-actin and total actin content was determined (fig. 2C); however, no increase of cellular G-actin content was observed after treatment of NIH 3T3 fibroblasts with lovastatin. This result prompted us to study the effects of various cytotoxins which are known to induce rounding up of cells by disruption of the actin cytoskeleton on cellular G-actin content. Similar to lovastatin no increase of G-actin was observed in cells treated with toxins that inactivate small GTPases of the Rho-subfamily either by ADP-ribosylation (C. botulinum exoenzyme C3, data not shown,C. limosum C3-like exoenzyme) or by glucosylation (C. difficile toxin B). A 50% increase of cellular G-actin content was observed after incubation with C. botulinum C2 toxin which ADP-ribosylates actin at Arg-177 (Aktories and Wegner, 1989). Total actin content after dissociation of F-actin with guanidine hydrochloride was not changed significantly by the bacterial toxins or lovastatin (data not shown). In all cases, cells were treated until rounding up of cells occurred (fig. 2C). No deleterious effects of the toxins or lovastatin on cell viability were found by trypan blue exclusion or determination of lactate dehydrogenase in the culture supernatant.
Because lovastatin prevents membrane association of microinjected recombinant Rho protein (Mohr et al., 1990), we studied the effect of this compound on distribution of endogenous Rho between membranes and cytosol in fibroblasts. Rho proteins were ADP-ribosylated with C. botulinum exoenzyme C3 in cytosol and membrane fractions of NIH 3T3 fibroblasts treated without or with lovastatin. As shown in figure 3 ADP-ribosylated Rho proteins increased in cytosol and decreased in membranes in a time- (fig. 3A) and concentration-dependent (fig. 3B) manner.

Effects of lovastatin on subcellular Rho protein distribution in NIH 3T3 cells after lovastatin treatment. (A) NIH 3T3 mouse fibroblasts were treated with lovastatin (30 μM) for the times indicated. Cells were lysed, separated into cytosol and membrane fraction and ADP-ribosylated with C. botulinum exoenzyme C3 as described under “Materials and Methods.” [32P]ADP-ribose incorporated into Rho proteins was quantitated with the phosphorimager software, and at time zero it was set to 100%. Mean values and the range of the radioactivity incorporated of two independent experiments are depicted in the upper panel. The lower panel shows the results of the phosphorimager analysis of the polyacrylamide gels. (B) Cells were incubated for 24 h with the concentrations of lovastatin given in the figure and were processed as described under “Materials and Methods.” Radioactivity incorporated into untreated cells was set to 100%. In the upper panel mean values and the range of [32P]ADP-ribose incorporated from two independent experiments are shown. The autoradiogram of polyacrylamide gels obtained with the phosphorimager is presented in the lower panel.
Effects of cycloheximide on morphology and RhoA content of NIH 3T3 fibroblasts.
Cycloheximide, a protein synthesis inhibitor, has been reported to prevent Swiss 3T3 cell rounding induced by compactin (Schmidt et al., 1982). Here we show that cycloheximide has similar effects on lovastatin-treated NIH 3T3 cells. Subconfluent cells were treated without (fig. 4A, upper panel) or with (fig. 4A, middle panel) lovastatin (30 μM) alone. Within 24 h lovastatin-treated cells rounded up (fig. 4A, middle panel). In the presence of lovastatin (30 μM) plus cycloheximide (5 μg/ml), cells remained flat (fig. 4A, lower panel). However, as compared with untreated cells (fig. 4A, upper panel), cell density of lovastatin- and cycloheximide-treated cells (fig. 4A, lower panel) was lower. Most likely cells have arrested growth in response to lovastatin and the protein synthesis inhibitor cycloheximide. When Swiss 3T3 cells preincubated with cycloheximide and lovastatin (fig. 4B, upper panel) were microinjected with C. botulinum exoenzyme C3 they assumed a round shape (fig. 4B, lower panel). Similarly, NIH 3T3 cells pretreated with lovastatin (30 μM) and cycloheximide (5 μg/ml) for 12 h (fig. 4C, upper panel) rounded up within 24 h afterC. botulinum exoenzyme C3 was added to the culture medium (fig. 4C, lower panel).

Effects of cycloheximide and C. botulinum exoenzyme C3 on lovastatin-treated NIH and Swiss 3T3 mouse fibroblasts. (A) NIH 3T3 cells were incubated without (upper panel) or with lovastatin (30 μM, middle panel) or lovastatin (30 μM) and cycloheximide (5 μg/ml, lower panel). Photomicrographs of cells are shown after 24 h treatment. Bar, 20 μm. (B) Swiss 3T3 cells were preincubated with cycloheximide (5 μg/ml) and lovastatin (30 μm) for 24 h (upper panel); thereafter, C. botulinum exoenzyme C3 (100 μg/ml) was microinjected and photographs were taken 1 h later. Bar, 5 μm. (C) NIH 3T3 cells were incubated with lovastatin (30 μM) and cycloheximide (5 μg/ml) for 12 h. Thereafter, C. botulinum exoenzyme C3 (50 μg/ml) was added to the culture medium; 24 h later photographs were taken from cells treated with lovastatin and cycloheximide only (upper panel) or cells incubated with lovastatin, cycloheximide and exoenzyme C3 (lower panel). Bar, 10 μm.
Effect of Val14RhoA on lovastatin-treated Swiss 3T3 cells.
If inactivation of Rho is essential for cell rounding by lovastatin, active Rho GTPase should revert the lovastatin phenotype. Therefore, we performed microinjection studies. Results of a typical experiment are shown in figure 5. Swiss 3T3 cells were treated with lovastatin (30 μM) for 24 h. Thereafter, dominant active Val14RhoA which is unable to hydrolyze GTP was microinjected into the cells. As can be seen in figure 5, the cell which was round before microinjection (upper panel) returned to a flat morphology after microinjection of Val14RhoA (lower panel).

Morphological effects of Val14RhoA on lovastatin-treated Swiss 3T3 cells. Lovastatin (30 μM) was added to culture medium of Swiss 3T3 cells for 24 h. Thereafter, Val14RhoA (1 mg/ml) was microinjected. The photomicrograph shows the same cell before (upper panel) and 1 h after microinjection of Val14RhoA (lower panel). Bar, 10 μm.
Effects of CNF1 and lovastatin treatment on NIH 3T3 cell morphology and exoenzyme C3 ADP-ribosylation of Rho proteins.
The cytotoxin CNF1 from E. coli is supposed to exert its effects by activation of Rho proteins (Oswald et al., 1994). To test whether CNF1 has effects similar to microinjected dominant active Val14RhoA, we added CNF1 to lovastatin-treated NIH 3T3 cells. As can be seen in figure 6A, NIH 3T3 cells rounded by lovastatin (upper panel) returned to a flat morphology within 6 h after CNF1 treatment (lower panel).

Effects of CNF1 on lovastatin-treated NIH 3T3 cells. (A) NIH 3T3 cells were incubated with lovastatin (30 μM) for 24 h (upper panel) and photographed. CNF1 lysate was added and 6 h later cells were photographed again (lower panel). Lysates ofE. coli transformed with vector only caused no morphological changes (not shown). Bar, 20 μm. (B) NIH 3T3 cells were treated without (Con) or with CNF1-containing E. colilysate (CNF1) or lovastatin (30 μM, Lova) or with lovastatin and CNF1 lysate (Lova + CNF1) for 24 h. Thereafter, cells were lysed, separated into cytosol (c) and membrane (m) fraction and ADP-ribosylated as described under “Materials and Methods.” Phosphorimager analysis of the radioactivity incorporated into Rho protein is shown.
We further studied the effect of CNF1 on Rho protein ADP-ribosylation in cytosol and membranes of NIH 3T3 cells. After lovastatin treatment the amount of Rho protein which was ADP-ribosylated by exoenzyme C3 increased in cytosol and decreased in membranes (fig. 6B). CNF1 caused an increase of ADP-ribosylated Rho in cytosol as well as in membranes. Combination of CNF1 with lovastatin caused an increase of Rho ADP-ribosylation in the cytosol and diminished Rho ADP-ribosylation in membranes as compared with controls. We also observed a shift of Rho protein to an apparently higher molecular mass in membranes of CNF1-treated NIH 3T3 cells.
Discussion
HMG-CoA inhibitors induce cell rounding and breakdown of the actin cytoskeleton in cultured cells (Schmidt et al., 1982; Fentonet al., 1992; Bifulco et al., 1993). Here we show for the first time that the rounding up of cells in response to lovastatin reduced the amount of F-actin but did not increase the amount of monomeric G-actin. A similar decrease in F-actin without apparent increase of G-actin was detected with various cytotoxins including C. botulinum exoenzyme C3, C. limosumC3-like exoenzyme and C. difficile toxin B. These toxins induce ADP-ribosylation [C3-like transferases (Braun et al., 1989)] or glucosylation [C. difficile toxin B (Just et al., 1995)] of Rho proteins. The lack of increase of G-actin content after C3-like toxin or C. difficile toxin B treatment is probably caused by the indirect effect of these cytotoxins on the actin cytoskeleton: depolymerization of F-actin by these agents does not change cellular concentrations of G-actin. Therefore, it is feasible that redistributed actin forms oligomers which are neither detected as F-actin bundles nor as G-actin. Alternatively, G-actin released from filaments by the toxins is sequestered by actin-binding proteins. In contrast, C. botulinum C2 toxin, which disrupts the cytoskeleton by direct ADP-ribosylation of G-actin (Aktories et al., 1986), decreased F-actin and increased the amount of G-actin as reported previously (Aktories and Wegner, 1989). The findings that lovastatin changes the G/F-actin ratio similar to the Rho-modifying toxins indicate that lovastatin acts more like these Rho-inactivating bacterial enzymes than like the actin-ADP-ribosylating C2 toxin.
Therefore, the effects of lovastatin on Rho-family proteins were studied in more detail. By C. botulinum exoenzyme C3 ADP-ribosylation, we observed a time- and concentration-dependent shift of Rho proteins from membrane to cytosol after lovastatin treatment. This may be explained by the fact that membrane binding of Rho proteins is no longer possible, when carboxy-terminal polyisoprenylation is prevented by lovastatin. Consequently, the amount of Rho proteins decreases in membranes and increases in cytosol. The redistribution of Rho observed is in accordance with previous findings of our laboratory. In Xenopus laevis oocytes polyisoprene-free recombinant Val14RhoA is polyisoprenylated within 30 min and translocates to the membrane. Lovastatin prevents membrane translocation of microinjected recombinant Val14RhoA (Mohr et al., 1990). However, apart from Rho proteins, other small GTP-binding proteins accumulate in the cytosol through lovastatin treatment; in insulin-secreting HIT-T15 cells, lovastatin increases concentrations of various cytosolic 20- to 30-kDa GTP-binding proteins (Li et al., 1993; Bhullar, 1996). Thus, not only Rho but also other small GTPases like Ha-Ras or Rab2 disappear from membranes and appear in the cytosol of cultivated cells after lovastatin treatment (Wei et al., 1992; Klinz,1994).
Cycloheximide has been shown to prevent lovastatin-induced rounding up of human renal carcinoma K1 cells (Fenton et al., 1992) and of thyroid epithelial FRTL cells (Bifulco et al., 1993). In their report, Fenton and co-workers (1992) proposed that prenylated proteins promote polymerization by functional inhibition of a short half-life protein whose normal activity is depolymerization of actin. If this protein is no longer synthesized in the presence of cycloheximide, depolymerization of F-actin cannot occur, not even in the presence of inhibitors of prenylation. We confirmed that cycloheximide inhibits lovastatin-induced rounding up of cells. However, our findings do not support the hypothesis of Fenton and co-workers: after addition of C3 to culture medium or microinjection of C3, lovastatin and cycloheximide-treated cells still rounded up. Thus, it appears that at least for the effects of C3 transferase no additional labile protein factor is necessary for depolymerization of F-actin.
Further evidence for the involvement of Rho in lovastatin-induced rounding up of cells is obtained from microinjection studies with constitutive active Rho. The dominant active Val14RhoA caused flattening of lovastatin-treated cells (fig. 5). Because the high amounts of microinjected recombinant protein may induce unspecific effects, we chose a second approach to study the possible involvement of Rho in effects of lovastatin on the cytoskeleton. It has been suggested that E. coli CNF1 and CNF2 toxins activate Rho proteins: CNF1 and CNF2 increase actin stress fiber formation in cultured cells (Falzano et al., 1993; Oswald et al., 1994) in a manner very much like microinjected dominant active Val14RhoA (Paterson et al., 1990). Indeed, it was recently shown that CNF1 posttranslationally modifies Rho proteins by deamidation of Gln63 to Glu63 (Schmidt et al., 1997). Gln63 forms part of the guanine nucleotide binding and hydrolyzing site. Conversion of Gln63 to Glu63 impairs the ability of Rho proteins to hydrolyze GTP to GDP and inorganic phosphate. Thereby Rho proteins are rendered permanently active. It is suggested that the CNF1-induced reversion to a flat morphology of lovastatin-treated NIH 3T3 cells is caused by this posttranslational activating modification of Rho proteins.
Activating Rho protein modifications (Val14RhoA, Glu63RhoA) seem to be sufficient to reestablish the actin cytoskeleton. Obviously, polyisoprenylation of Rho proteins for membrane binding is not necessary because even in the presence of lovastatin cells revert to a flat morphology through CNF1. Also, a concomitant increase in membrane binding through overexpression or microinjection of large amounts of Rho proteins is not necessary: As in figure 6B, CNF1 does not raise membrane concentrations of Rho proteins in lovastatin-treated NIH 3T3 cells. Figure 6B also shows that apart from causing a small retention in Rho protein migration in SDS-PAGE, CNF1 increases the amount of Rho protein available for ADP-ribosylation. In addition to posttranslational modification, increase of Rho protein concentrations may cause the cells to return to a flat morphology.
As to the type of Rho protein that is responsible for maintaining or reestablishing actin cytoskeleton organization, one must consider that microinjection of a large amount of active RhoA might mimic functions of Rac or Cdc42 proteins. CNF1 also has been found to modify RhoA, Rac and Cdc42. However, the findings with Val14RhoA and the CNF1 substrate specificity suggest that Rho-subtype proteins are sufficient for reconstitution of the flat morphology of lovastatin-treated cells. In summary, the results presented, for the first time, give direct evidence for the involvement of Rho-subfamily proteins in the disruption of the actin cytoskeleton induced by the HMG-CoA inhibitor lovastatin in cultured cells.
Acknowledgments
We thank B. Neufang for expert technical assistance. We thank I. Just, University of Freiburg, for advice on determination of cellular G-actin content. Also, we acknowledge that, while these studies were in progress, Dr. M. Sugai, Uniformed Services University, Bethesda, MD, reported the effects of CNF2 on lovastatin-treated cells at the Gordon Conference on Microbial Toxins and Pathogenesis (July14–19, 1996).
Footnotes
-
Send reprint requests to: Dr. K. Aktories, Institut für Pharmakologie und Toxikologie, Universität Freiburg, Hermann-Herder-Str. 5, D-79104 Freiburg, Germany.
-
↵1 This work was supported by the Deutsche Forschungsgemeinschaft.
-
↵2 Present address: Psychiatrische Klinik Rheinau, CH-8462 Rheinau, Switzerland.
- Abbreviations:
- CNF1 and 2
- Escherichia colicytotoxic necrotizing factor 1 and 2
- F-actin
- filamentous actin
- G-actin
- globular actin
- GTPase
- guanosine 5′-triphosphate hydrolase
- HMG-CoA
- hydroxymethylglutaryl coenzyme A
- MVA
- mevalonic acid
- NAD
- nicotinamide adenine dinucleotide
- NBD-phalloidin
- (7-nitrobenz-2-oxa-1,3-diazol-4-yl)phalloidin
- RhoGDI
- Rho guanosine nucleotide dissociation inhibitor
- SDS-PAGE
- sodium dodecyl sulfate-polyacrylamide gel electrophoresis
- S.E.M.
- standard error of the mean
- Received February 10, 1997.
- Accepted July 7, 1997.
- The American Society for Pharmacology and Experimental Therapeutics




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}