Article Text

Abstract
BACKGROUND Inactivation of the tumour suppressor gene p16 (CDKN2/MTS-1/INK4A) and K-ras mutations are among the most frequent genetic alterations in human malignancies.
AIMS To investigate the tumour suppressor gene p16 and its possible association with K-ras mutations in intrahepatic cholangiocarcinomas of the liver.
METHODS The status of p16 was evaluated in 41 cholangiocarcinomas by methylation specific polymerase chain reaction, microsatellite analysis, DNA sequencing, and immunohistochemical staining. K-rasmutations were determined by direct DNA sequencing analyses after microdissection. The results obtained were correlated with histopathological variables and patient survival.
RESULTS Hypermethylation of the 5′ CpG island of the p16 gene was found in 34 of 41 (83%) carcinomas. Homozygous deletion at the p16 region was present in two (5%), and loss of heterozygosity (LOH) in eight cases (20%). We failed to detect p16 gene missense mutations. K-rasmutations were found in 22 of 41 (54%) cholangiocarcinomas and in two cases of tumour surrounding non-neoplastic liver tissue. All 22 cancers with K-ras mutations also exhibited methylated p16. We failed to observe a correlation between K-ras or p16 status and histopathological factors or prognosis of patients.
CONCLUSION These data suggest that inactivation of the p16 gene is a frequent event in cholangiocarcinoma. The most common somatic alteration is promotor methylation of the p16 gene which is closely associated with K-ras mutations. We failed to establish p16 or K-ras status as independent prognostic factors in these tumours.
- cholangiocarcinoma
- p16
- K-ras
- histopathology
- prognosis
- methylation
Abbreviations used in this paper
- CCC
- cholangiocarcinoma
- HCC
- hepatocellular carcinoma
- PCR
- polymerase chain reaction
- MSP
- methylation specific PCR
- LOH
- loss of heterozygosity
- SSCP
- single strand conformation polymorphism
Statistics from Altmetric.com
Intrahepatic cholangiocarcinoma (CCC) is usually a fatal malignant neoplasm originating from biliary epithelia or cholangiocytes and constitutes about 5% of primary liver cancers.1 At present, the cellular and molecular mechanisms leading to oncogenesis of cholangiocytes are unclear. There is increasing evidence that carcinogenesis must be understood in terms of accumulation of mutations in regulatory genes, including activation of oncogenes and inactivation or loss of tumour suppressor genes.2 ,3 Among the available candidates, the K-ras oncogene may be involved in cholang- iocarcinogenesis.4 Theras gene product has a key role in controlling cell growth and differentiation through its intrinsic GTPase activity.5 Several lines of evidence have shown that the cell cycle machinery, specifically the circuit cyclin D1/cyclin dependent kinase-p16-pRb, lies downstream ofras.5 Point mutations that activate the ras protein and its downstream cascade have been observed in human tumours.6 The tumour suppressor gene p16 (INK4A/MTS-1/CDKN2) is believed to encode a negative regulatory protein that regulates the progression of eucaryotic cells through the G1 phase of the cell cycle.7 Recent data indicate that inactivation of p16 in hepatocellular carcinoma (HCC) is due to de novo hypermethylation of the 5′ promotor associated CpG island.8-10 Introduction ofras mutations into cells deficient in p16 is sufficient to induce characteristics of cellular transformation such as tumour formation in vivo. Data from K-rastransformation studies suggest that the mutated K-ras oncogene induces p16 hypermethylation and consequently inactivation of the p16 gene.11 It was also reported that H-ras overexpression increased DNA methyltransferase activity, suggesting a possible link between ras and (p16) methylation.12
To date, no study has simultaneously assessed K-rasmutations and p16 alterations in CCC of the liver. Hence we analysed the status of p16 and prevalence of K-ras oncogene mutations in these tumours. Data were compared with histopathological results and with the prognosis of patients.
Materials and methods
PATIENTS AND TISSUE SAMPLES
Forty one patients with CCC undergoing partial hepatectomy (segmental or lobar resection) between 1994 and 1997 were included in this retrospective study. No patient received preoperative or adjuvant chemotheraphy or radiotherapy. All patients underwent surgery of curative intent (R0 resections). Patients who underwent orthotopic liver transplantation were excluded from the study.
Each tumour was re-evaluated with regard to typing, staging, and grading (WHO 199413). Tumour typing and staging were performed using WHO and UICC (1997)14 criteria, respectively. In addition, every tumour was examined macro- and microscopically for the presence of vascular invasion, satellites, multiplicity, inflammatory reaction, necrosis, and dysplasia in the surrounding liver tissue and cirrhosis. In all cases, slides prepared from four different paraffin blocks of tissue, sampled from different tumour areas, were examined. Pathohistological data are summarised in table 1.
Patients and pathohistological data
DNA SAMPLES
For each CCC sample, histopathological lesions of interest were first identified on routinely stained slides. Parallel sections were cut with the microtome set at 6 μm, and the slides dried overnight at 37°C. Corresponding areas of interest were delineated and microdissected after rapid staining with haematoxylin and eosin. Thereafter the tissue was scraped off the slide (the sections were covered with 25 μl of Tris buffer, 0.05 mol) with the tip of a sealed glass pipette and then sucked into a microcapillary. Tissue samples were placed in Eppendorf tubes and incubated with proteinase K at 37°C overnight. Proteinase K activity was inactivated by heating to 95°C for 10 minutes, and the resulting solutions were used directly as templates for K-ras analyses. For DNA extraction, standard methods were used: after incubation with proteinase K at 37°C overnight the tissue was extracted twice in phenol and twice in chloroform, followed by ethanol precipitation.
K-rasMUTATION ANALYSIS
All pre-polymerase chain reaction (PCR) tissue was handled in an environment free of PCR products. All samples were coded and the investigator was blinded to patient clinical details. Deparaffinised tissue was recovered by a 15 minute incubation with xylene followed by centrifugation for five minutes at 14 000 rpm. This was repeated. The tissue pellet was washed twice in absolute ethanol followed by two washes in phosphate buffered saline. The pellet was incubated with 10 pellet volumes (approximately 500 μl) of lysis buffer (0.32 M sucrose, 10 mM Tris HCl, 1% (v/v) Triton X-100), and 0.2 volumes of proteinase K (final concentration 400 μg/ml) for 2–3 days at 37°C. DNA was phenol-chloroform extracted and precipitated in ethanol using conventional techniques. The resulting DNA pellet was resuspended in 50 μl of TE buffer, pH 7.4 (10 mM Tris HCl, pH 7.4, 1 mM EDTA, pH 8.0). DNA samples were stored at −20°C.
The first exon of K-ras was amplified by PCR using primers designed to avoid amplification of the K-ras pseudogene. The primers used were 5′-ATTATAAGGCCTGCTGAAAATG- ACTGA-3′ (upstream primer) and 5′- ATATGCATATTAAAACAAGATTTACCT- CTA -3′ (downstream primer) giving a 155 base pair product. Amplification was performed using a touchdown PCR technique15 ,16 from 63 to 53°C over 10 cycles, followed by 30 cycles at 94°C, 53°C, and 72°C.
PCR products were purified using the Qiaquick PCR purification kit (Qiagen, Hilden, Germany) and sequenced using dye primer cycle sequencing and AmpliTaq polymerase FS on an Applied Biosystems 373 DNA sequencer (ABI 373; Applied Biosystems-Perkin-Elmer/Cetus, Norwalk, Connecticut, USA).
CONTROLS
DNA from colon carcinoma cell lines SW480 (Clontech, Palo Alto, California, USA) and HCT116 (American Type Culture Collection, ATCC, Rockville, Maryland, USA) with known K-rasmutations at codon 12 (GTT) and codon 13 (GAC), respectively, were used as positive controls in each of the parallel procedures. Negative controls, without DNA, were run as controls for contamination.
If a mutation was detected, it was confirmed by amplification and sequencing of a fresh DNA sample using the upstream primer. Any sequences which proved difficult to read were re-amplified and re-sequenced.
METHYLATION SPECIFIC PCR (MSP) OF THE p16 GENE
The CpG WIZ p16 methylation assay kit was used (Oncor Inc, Gaithersburg, Maryland, USA) in accordance with the manufacturer's instructions. After an initial bisulphide reaction to modify DNA, PCR amplification with specific primers was performed to distinguish methylated from unmethylated DNA (unmethylated p16 primers: 5′ - TTATTAGAGGGTGGGGTGGATTGT - 3′, 5′-CAACCCCAAACCACAACCATAA-3′); methylated p16 primers: 5′-TTATTA GAGGGTGGGGCGGATCGC-3′, 5′-GAC CCCGAA CCGCGACCGTAA-3′). DNA (7 μg/100 μl) was denatured by 0.2 M NaOH for 10 minutes at room temperature. DNA Modification Reagent I was added, incubated for 24 hours at 50°C, and subsequently purified by DNA Modification Reagents II and III in the presence of 50 μl of water. The bisulphate modification of DNA was completed with 0.3 M NaOH treatment for five minutes, followed by ethanol precipitation. For hot start PCR, the PCR mixture contained Universal PCR buffers (1X9, 4dNTPs (1.25 nM)), U or M primers (300 ng each per reaction). Annealing temperature was 65°C for 35 cycles. The PCR product was directly electrophoresed on a 3% agarose gel, stained with ethidium bromide, and visualised under UV illumination. Bisulphite converted DNA from corresponding normal liver tissue from each patient served as a negative control, as indicated by the presence of the unmethylated but not the methylated band.
MICROSATELLITE ANALYSIS OF THE LOSS OF HETEROZYGOSITY (LOH) AND DNA SEQUENCING
We used nine microsatellite markers flanking the chromosome 9p21 region were the p16 gene is located. The markers used were D9S161, D9S126, D9S171, D9S1752, D9S1748, D9S1747, D9S1749, D9S1751, and IFNA and were obtained from Research Genetics (Hubtsville, Alabama, USA). The primers were labelled with 32P ATP. PCR amplification were performed in a 10 μl reaction volume including 30 ng of genomic DNA, 10 mM Tris HCl, 50 mM KCl, 1.5 mM MgCl2, 62.5 μM deoxynucleotide triphosphate, 0.1 U Taq DNA polymerase, and 1 pmol of each primer. A total of 45 cycles were performed with annealing temperatures of 53° and 55°C. PCR products were subsequently electrophoresed, dried, and autoradiographed. Homozygous deletion of the p16 gene was confirmed by comparative PCR involving amplification of two different sets of primer pairs in the same reaction mixture. For homozygous deletion, the control marker D9S126 was used. Homozygous deletion was scored if the signal intensity (assessed by visual examination and densitometer) of the p16 in tumour tissues was at least 10-fold less than the signal from the non-tumorous tissues. The intensity of the D9S126 control allele was approximately equal in tumour and corresponding non-tumorous liver.
For mutation analysis on exon 1,2 (2A, 2B, and 2C) we performed SSCP analysis using the primers described by Hussussian and colleagues.17 Single strand conformation polymorphism (SSCP) analysis was performed as described in detail previously.18 The primers were labelled with32P ATP and each sample was subjected to PCR analysis (denaturing for 30 seconds, annealing for 45 seconds, extension for 30 seconds at 94°C, 55–60°C, and 72°C, respectively). The PCR products were electrophoresed, the gels dried, and autoradiographed. Variant SSCP bands were cut out from the gel and the DNA eluted. Variant bands and 3 μl of the eluted DNA were used as templates for unlabelled PCR. After purification of the PCR products, sequencing analysis was performed using the DNA Sequenase Kit (Amersham, Germany) and an automatic sequencing analyser (ABI 373; Applied Biosystems-Perkin-Elmer, Germany). All mutations were confirmed by direct sequencing of the amplified tumour and corresponding non-tumorous DNA to identify germline mutations and polymorphisms. The sequences of all primers used for amplification are available from the authors on request.
IMMUNOHISTOCHEMICAL ANALYSIS AND ASSESSMENT
Immunohistochemical analysis was performed as described previously.19 In all cases, tumour and non-neoplastic liver tissue were examined.
The following antibodies were used: ras (ras, monoclonal Clone F132; mouse, dilution 1:120; final concentration 10 μg/ml; Boehringer Mannheim, Mannheim, Germany), p16 (polyclonal; rabbit, dilution 1:500; Pharmingen, San Diego, California, USA).
Sections known to stain positively were included in each batch and negative controls were also performed by replacing the primary antibody with mouse or goat ascites fluid (Sigma-Aldrich Biochemicals, St Louis, Missouri, USA).
To minimise interobserver error, all counts were performed separately. In three cases, in which conflicting numbers of positive cells were evaluated, recounting was performed to obtain consensus.
STATISTICS
Differences in frequencies between subgroups were analysed using the Kruskal-Wallis test and the Mann-Whitney U test for unpaired samples. Correlation coefficients were calculated according to Pearson, and χ2 statistics were used for contingency tables. Overall observed survival functions and probabilities were estimated using the Kaplan-Meier method. The log rank test was used to detect differences between survival curves for stratified variables. Identification of relevant prognostic factors was performed with univariate Cox regression analyses. The significance level was defined as p<0.05.
Median follow up of our patients was 210 days (range 50–1749 days). No patient was lost to follow up.
The medical records of all 41 patients were re-examined to assess the status of disease at the closing date of the study (30 April 1998). At this time, eight patients were still alive. All patients who died during the follow up period had intrahepatic and metastatic disease on their last visit to the oncological outpatients clinic. We concluded that death in these patients was related to CCC.
Median survival was 210 days (95% confidence interval 156, 264 days) and the one year survival rate was 31% (95% confidence interval 17, 45 days).
Results
HISTOPATHOLOGICAL FEATURES
Pathological data are summarised in table 1.
METHYLATION OF THE p16 PROMOTOR
Methylation specific PCR revealed that 34 of 41 carcinomas showed aberrant methylation at the 5′CpG island of the p16 gene (tables 2, 3; fig 1). Despite microdissection, amplification of unmethylated templates was detected to some degree (fig 1C), probably because of contaminated normal intratumorous tissue (fibroblasts, endothelial cells, inflammatory cells). In corresponding non-tumorous liver tissue, methylated templates were amplified in one case. This patient exhibited a high degree of inflammation.
K-ras mutation and p16 methylation according to stage and grade of disease
K-ras mutation and p16 methylation in cholangiocarcinoma
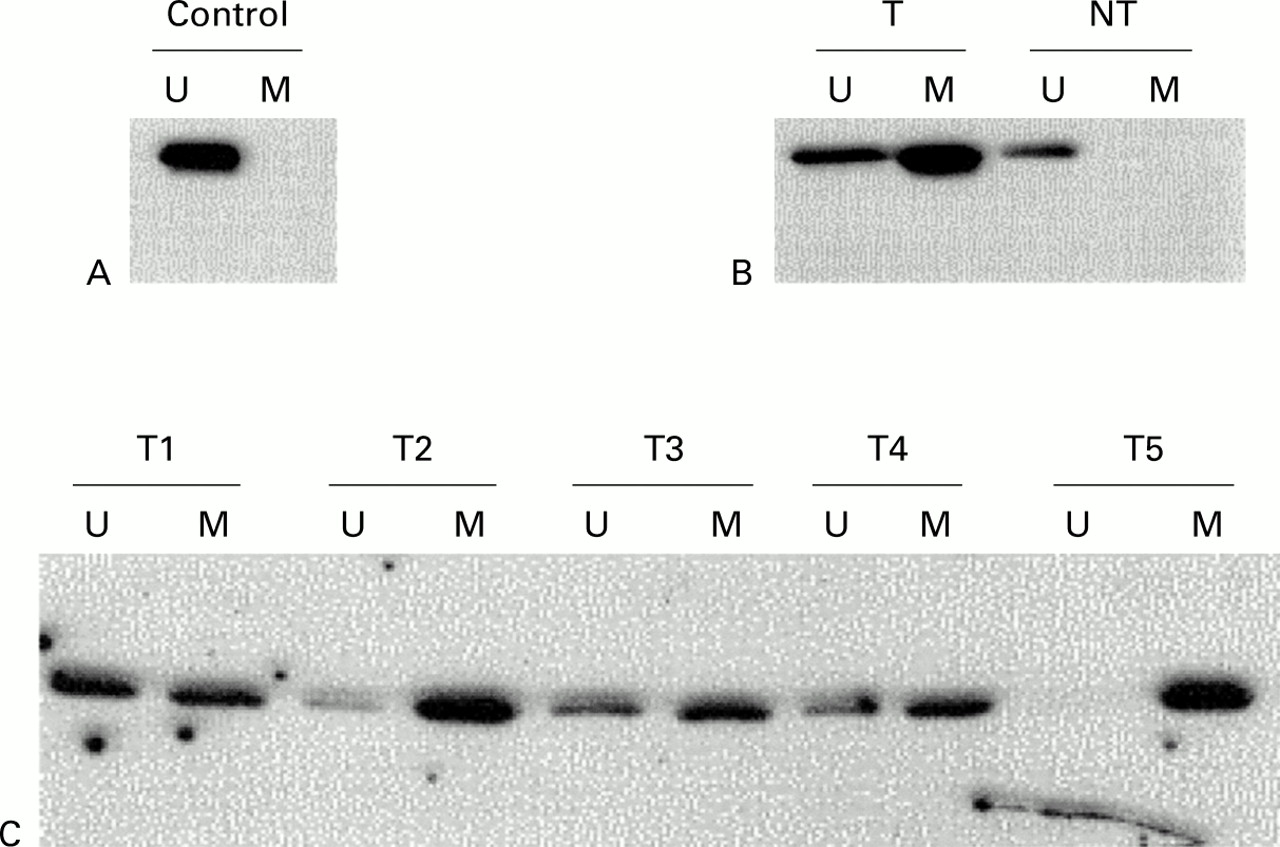
Methylation analysis of p16 in cholangiocarcinoma. Methylation specific PCR (MSP) results are expressed as unmethylated p16 specific bands (U) or methylated p16 specific bands (M). (A) Bisulphite converted DNA from normal liver tissue (N) served as a negative control as indicated by the presence of the U but not the M band. (B) MSP results of case No 28. The tumour surrounding non-neoplastic liver tissue (NT) with unmethylated p16. (C) Representative cases of cholangiocarcinoma. The numbers of the cholangiocarcinoma cases are shown above the corresponding lanes.
Of the 22 cancers with K-ras mutations, all specimens also showed methylated p16 (table 3). All 34 cases with aberrant methylation of the p16 gene showed complete loss of immunoreactivity within the tumour tissue (fig 2A). In the seven cases shown by MSP to lack p16 promotor methylation, nuclear staining of p16 protein was observed in nearly all tumour cells with a moderate to strong intensity of immunoreactivity (fig 2B). In normal liver tissue, p16 protein was detected in all cases. Comparing the status of p16 with histopathological variables, we failed to observe a significant relation. Methylation of p16 did not correlate with stage or grade or with the mutation pattern of K-ras.

Immunostaining of p16 protein in cholangiocarcinoma. (A) Patient No 28 (same patient as in figure 1B) with a methylated p16 gene. Complete loss of p16 expression. (B) Immunohistochemical staining of p16 in a moderately differentiated cholangiocarcinoma without p16 methylation. Strong nuclear positivity of the tumour cells (brown reaction product). (Original magnification ×40.)
LOH ON CHROMOSOME 9p21 AND HOMOZYGOUS DELETION
Eight of 41 carcinomas (20%) showed LOH in at least one locus on chromosome 9p21. The highest frequency of LOH was observed with the microsatellite marker D9S 1751 (seven cases) and the lowest with D9S171 (one case). Homozygous deletion of the p16 gene was observed in two cases. In these tumours, the signal for the alleles at p16 was highly reduced whereas the control marker D9S126 showed similar intensity in both tumour and non-tumorous liver tissue. Immunohistochemistry showed that there was no specific staining in cases with homozygous deletion of the p16 gene.
We failed to detect mutations of the p16 gene. In SSCP analysis, a mobility shift was detected in one carcinoma, but we did not find a specific mutation of p16 in this tumour.
K-rasSTATUS
PCR amplification and DNA sequencing enabled detection of heterozygous mutations in 22/41 CCCs (54%). Seventeen patients had a mutation of codon 12 and five of codon 13. Eight of 22 mutations were G→A transitions. No patient had multiple mutations. In two cases, mutation of the K-ras gene was detected in non-neoplastic tumour surrounding liver tissue (table 3). One mutation was at codon 12, the other at codon 13. The base pair changes in non-neoplastic tissue consisted of a G→A transition, producing an amino acid substitution of glycine for aspartic acid.
We failed to observe an association between K-ras mutation pattern and histopathological variables.
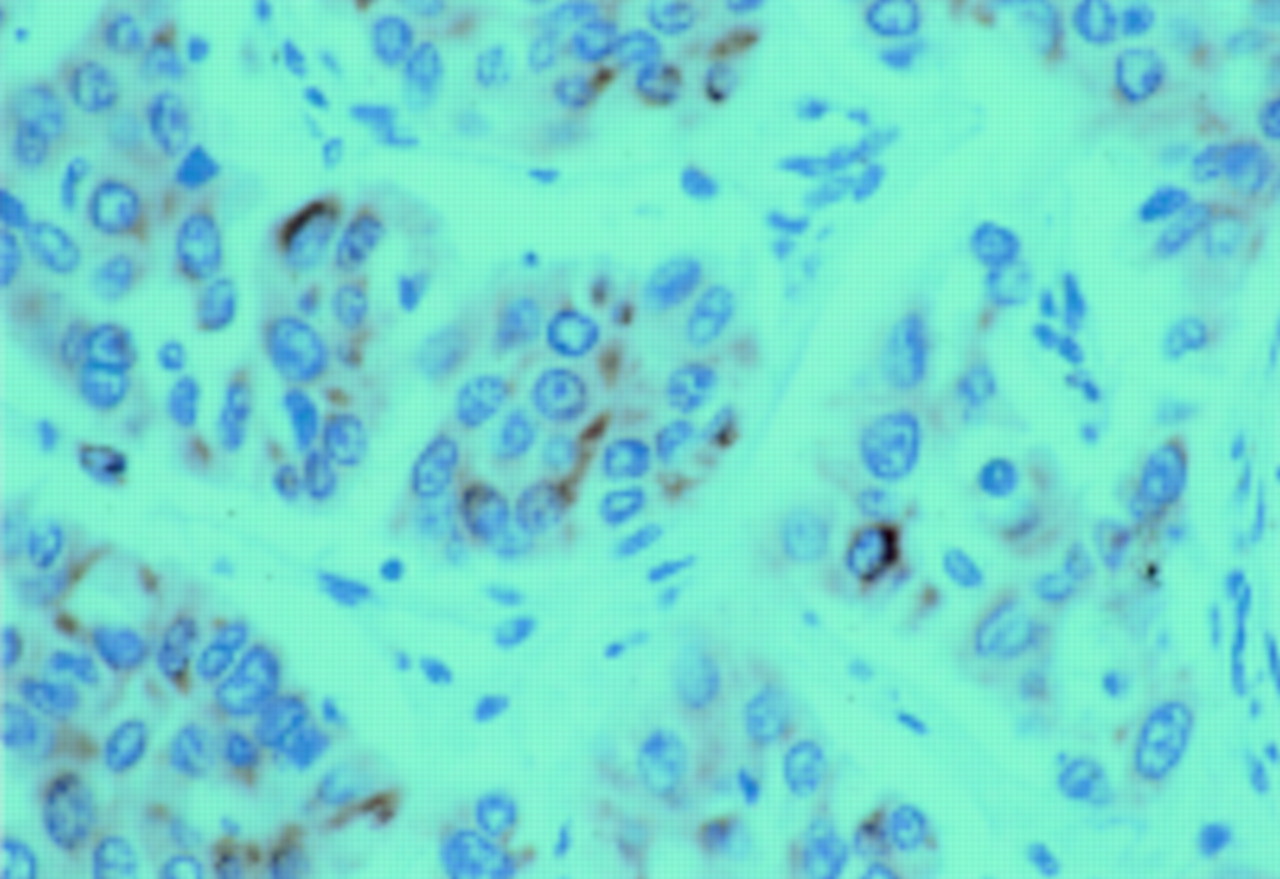
When we performed a semiquantitative assessment of p21rasimmunoreactivity (fig 3), neither the staining intensity nor the number of positive cells correlated with the mutation pattern or with other histopathological parameters (table 3). Non-neoplastic liver tissue was occasionally positive for p21ras.
{kind=link}
{kind=link}
{kind=link}
Immunostaining of the ras protein with perinuclear staining (red reaction product) of tumour cells. (Original magnification ×60.)
SURVIVAL RATE
Survival analysis took into account the following variables: K-ras and p16 status (mutated, methylatedv wild type, unmethylated), UICC tumour stage (UICC 1997), grading, vascular invasion, multiplicity, satellites, dysplasia, inflammatory reaction, necrosis, and patient age.
As expected, UICC stage, extent of the primary tumour (pT category), presence of lymph node metastases (pN category), and histological grade of tumour differentiation were significant prognostic parameters in univariate analysis. Neither p16 nor K-raswere related to the prognosis of our patients. The odds ratios for all factors examined are given in table 1. On multivariate analysis, only extent of primary tumour (pT category) and lymph node status (pN category) had an independent prognostic impact.
Discussion
In our study, we examined the frequency of mutations of the K-ras oncogene and the status of p16, the most frequent genetic aberrations in human epithelial tumours. Whereas p16 was examined in HCCs,8 ,9 data are still lacking on p16 in CCCs of the liver, the second commonest primary liver tumour. Our study showed that the p16 tumour suppressor gene was inactivated in a high percentage of CCCs. p16 inactivation occurred via different mechanisms, including methylation and homozygous deletion. We did not detect specific point mutations of the p16 gene. The most frequent mechanism of inactivation in our series was transcriptional loss due to DNA methylation at a CpG island in the promotor region, which was detected in 83% of all carcinomas. In all tumours without detectable p16 protein, either CpG methylation or homozygous deletion was observed. For HCC, a high CpG methylation rate of 63% was reported recently, indicating that the main mechanism of inactivation of p16 in liver tumours is transcriptional loss due to DNA methylation at the 5′ promotor associated CpG island.8-10 In CCC, our findings indicate that the same mechanism could be responsible for inactivation of p16 in a high percentage of cases.
Despite using microscopic microdissection techniques to ensure examination of tumour free liver tissue, we observed weak amplification of methylated DNA in one case of tumour surrounding liver tissue. This weak signal of methylated sequence in non-tumorous tissue could indicate that de novo methylation may occur in premalignant or early subcellular changes in these cases.
In our series, we also looked for mutation and deletion of the p16 gene. However, homozygous deletion occurred in only two cases. To date, there are no consistent data on the frequency of genetic changes on chromosome 9p21 in intrahepatic CCC. The reported data from HCCs, however, indicate that homozygous deletions and mutations of the p16 gene are rare events in these tumours.10 A recent study, focussing on 10 patients with primary sclerosing cholangitis associatedextrahepatic CCC indicated that loss of chromosome 9p21 and inactivation of the p16 tumour suppressor gene are frequently observed in these patients.20
Mutations of the K-ras oncogene occurred in 54% of the CCCs examined, which is in agreement with the literature.4 ,21 In our study, we found a high prevalence of G→A transitions within codon 12 and also in codon 13. Both K-ras mutations that occurred in non-neoplastic liver tissue were G→A transitions. However, we failed to observe a correlation between the mutation pattern and histopathological variables of the tumour or prognosis of the patient. In our series, the status of K-ras or presence of activating mutations in this gene did not predict biological behaviour, as reported for other tumours (for example, colon cancer22-24). However, due to selection criteria (only cases with primary curative (R0) resection were examined), only a limited number of cases were assessed for our study. Therefore, the actual prognostic value of p16 and K-rasshould be examined in a larger group of patients.
Mutated ras is known to transform most immortal cells.25 ,26 Although the growth dysregulating events of the ras oncogene are still poorly understood, data from K-ras transformation studies suggest that activated K-raspromotes p16 methylation.27 Stable transformation of colon cancer cells with K-ras increased DNA methyltransferase activity, methylated the p16 gene, and suppressed expression of p16. Furthermore, it has recently been proposed that introduction of ras mutations into cells deficient in tumour suppressor genes such as p16 is sufficient to induce characteristics of cellular transformation such as anchorage independent growth and tumour formation in vivo.11 Studies on immortalised cells have shown that p16 suppressedras induced proliferation by blocking entry into the S phase of the cell cycle.28 ,29 One may speculate that in the case of inactivated p16, the transformed cell may lose its growth inhibitory factors, allowing the transformed cell to proliferate. Therefore, the presence of additional genetic changes, such as loss of the tumour suppressor gene p16, may enhance the carcinogenic potential of K-ras.
In conclusion, our data suggest that CpG methylation appears to be the main cause of inactivation of p16 in CCC. The overall frequency of p16 alterations detected, including deletion and methylation, was 88%. Silencing of p16—a critical regulator of cell cycle progression—is therefore one of the most frequent genetic defects in CCC. Our findings of a strong correlation between p16 methylation and K-ras mutation support the hypothesis of a close molecular link between K-ras and p16 in the pathogenesis of CCC.
Abbreviations used in this paper
- CCC
- cholangiocarcinoma
- HCC
- hepatocellular carcinoma
- PCR
- polymerase chain reaction
- MSP
- methylation specific PCR
- LOH
- loss of heterozygosity
- SSCP
- single strand conformation polymorphism