Article Text

Abstract
Aims—Despite its well established tropism for B cells, the nature of the cellular compartment(s) mediating primary and persistent Epstein-Barr virus (EBV) infection is still a matter of controversy. In view of the association of EBV with several lymphoid and epithelial malignancies, resolution of this issue is important.
Methods—Desquamated oropharyngeal epithelial cells from 10 patients with acute infectious mononucleosis and from seven chronic virus carriers were studied for evidence of EBV infection using in situ hybridisation for the detection of the small EBV encoded RNAs (EBERs) and of the viral genome. In addition, immunocytochemistry was used to detect the BZLF1 transactivator protein of EBV.
Results—There was no evidence of latent or replicative EBV infection in oropharyngeal epithelial cells in any of the samples. In contrast, EBV infected B cells were readily identified in a tonsil from a patient with infectious mononucleosis.
Conclusions—The results suggest that oropharyngeal epithelial cells are not a major site of EBV infection and provide further support for the notion that B cells mediate primary and persistent EBV infection.
- Epstein-Barr virus
- infectious mononucleosis
- B cells
- epithelial cells
- in situ hybridisation
Statistics from Altmetric.com
The Epstein-Barr virus (EBV) is a B lymphotropic herpes virus that infects over 90% of the adult human population worldwide. In vitro, EBV can infect resting B cells, transforming them into immortalised lymphoblastoid cell lines (LCLs).1 EBV is associated with several lymphoid neoplasms such as Burkitt's lymphoma, Hodgkin's disease, and post-transplant lymphoproliferative disorders.1 However, the tumour that has the most consistent association with the virus is an epithelial neoplasm, nasopharyngeal carcinoma.2
EBV infection of LCLs and of human tumours is primarily latent—infectious virus is usually not produced and expression of the viral genome is restricted to a limited set of so called “latent” proteins.1 EBV can establish at least three forms of latent infection characterised by variable expression patterns of these latent proteins.3 In all forms of EBV latency, the small EBV encoded non-polyadenylated nuclear RNAs, EBER-1 and EBER-2, are abundantly expressed.1 This makes the EBERs an ideal target for the detection of latently infected cells by in situ hybridisation. The switch from latent to lytic infection is usually heralded by the synthesis of the BZLF1 transactivator protein, which is followed by the expression of late viral genes and the replication of the viral genome.4
Most individuals undergo asymptomatic primary infection with EBV early in childhood.5 Delayed primary infection can be associated with the development of infectious mononucleosis, an EBV driven self limiting lymphoproliferative disorder.5 In either case, primary infection is followed by life long, mostly asymptomatic, persistent infection. Despite its well established tropism for B cells in vitro, the nature of the cellular compartment(s) mediating primary and persistent EBV infection is still a matter of controversy. It is well known that during primary, as well as in persistent EBV infection, infectious virus can be shed into the saliva.1 Using in situ hybridisation, the detection of the virus has been reported in desquamated oropharyngeal epithelial cells in throat washings from patients with infectious mononucleosis and from chronic virus carriers.6,7 Furthermore, EBV replication has been demonstrated in the upper epithelial cell layers of oral hairy leukoplakia, an AIDS associated epithelial lesion of the tongue.8,9 On the basis of these results, it has been suggested that oropharyngeal epithelial cells are the primary target for EBV infection.10 According to this model, the virus would persist in the epithelial cell compartment. Differentiation dependent EBV replication in epithelial cells would allow shedding of infectious virus into the saliva as well as secondary infection of B cells.10 There is, however, increasing evidence pointing to B cells as the main site of primary EBV infection and of virus persistence. EBV infection can be eliminated from bone marrow transplant recipients whose resident haemopoietic tissue has been destroyed.11 Furthermore, antiviral treatment of patients with infectious mononucleosis and of chronic carriers has been shown to reduce EBV shedding into the oropharynx, but not the levels of EBV carrying B cells in the peripheral blood.12,13 In addition, although EBV replication can be detected in the differentiated cell layers of oral hairy leukoplakia, latent infection of basal epithelial cells has not been demonstrated.9,14,15 In tonsils from patients with infectious mononucleosis, latently infected B cells are easily detectable, whereas latent EBV infection of epithelial cells has not been demonstrated convincingly.16–18 Moreover, recent studies have also suggested that B cells undergoing plasmacytoid differentiation might support lytic EBV infection and thus may represent a cellular source of the infectious virus detected in the saliva.16,18
To clarify this issue, we decided to repeat earlier studies of throat wash samples using a wider spectrum of in situ hybridisation and immunohistochemical methods.
Materials and methods
COLLECTION OF THROAT WASHINGS
Ten patients with serologically confirmed EBV associated infectious mononucleosis were included in our study. Throat washings were collected by gargling with 10 ml of phosphate buffered saline (PBS) and cells were pelleted. The cell pellets were resuspended and fixed in 10 ml formalin for 20 minutes. The cells were centrifuged again and the supernatants were removed by aspiration and discarded. The cell pellets were embedded in paraffin wax using the Shandon cytoblock kit in accordance with the manufacturer's instructions (Shandon, Astmoor Runcorn, UK). Supernatants from five patients with infectious mononucleosis were tested for the presence of infectious virus by an in vitro transformation assay. This confirmed shedding of infectious EBV in all cases. Throat washings from eight healthy volunteers were also processed as described. Seven of these individuals were confirmed by serology to be chronic EBV carriers, and one was EBV negative. To ensure adequate sampling for subsequent analysis, the cytoblocks were cut at different levels, with at least three sections from different levels mounted on each slide. For control purposes, an EBV positive LCL, B95.8, and an EBV negative Burkitt lymphoma cell line, Ramos, were also processed into paraffin wax blocks and sectioned. Furthermore, a tonsil from a patient with acute mononucleosis from an earlier study was investigated.18
IMMUNOCYTOCHEMISTRY
Paraffin wax embedded sections were dewaxed, rehydrated, and subjected to microwave irradiation in one litre of 0.01 M citrate buffer (pH 6.0) in a 750 W domestic microwave oven. The sections were cooled under running tap water and then stained using the alkaline phosphatase anti-alkaline phosphatase (APAAP) method. The nature of the desquamated cells was ascertained using the monoclonal antibodies KsPan1–8 against cytokeratins (Boehringer, Mannheim, Germany), C3D-1 (CD15), L26 (CD20), HM57 (CD79a), and a CD3 specific polyclonal rabbit antiserum (all from DAKO, High Wycombe, UK). The monoclonal antibody BZ-1 (DAKO), which is specific for the EBV encoded BZLF1 transactivator protein, was used for the detection of lytic EBV infection.
PLASMIDS AND PROBES
The plasmids pBSJJJ1 and pBSJJJ2 harboured inserts specific for EBER-1 and EBER-2, respectively.9 The plasmid pBSW contained the BamHI W fragment of the EBV genome.9 The plasmid pBSU6 containing an insert specific for the cellular U6 RNA was a kind gift from Dr R Ambinder, Baltimore, Maryland, USA. 35S labelled RNA probes were generated from these plasmids by in vitro transcription. Briefly, plasmid DNA was linearised and sense and antisense probes were generated in reactions that contained 1 mg of linearised plasmid DNA, 0.5 mM each of CTP, ATP, and GTP (Boehringer), 50 μCi 35S-UTP, 20 units of RNase inhibitor (Boehringer), 10 mM dithiothreitol (DTT), and 20 units of T7 or T3 RNA polymerase as required (Boehringer). For in situ hybridisation, the EBER specific antisense probes were combined to increase sensitivity. The sense probes transcribed from the plasmids pBSJJJ1 and pBSJJJ2 were also combined.
Cot1 DNA was obtained from Boehringer and labelled using an oligonucleotide 3′ end labelling kit with digoxigenin labelled ddUTP (Boehringer) according to the manufacturer's instructions.
IN SITU HYBRIDISATION AND DOUBLE LABELLING.
For RNA–RNA in situ hybridisation, sections were dewaxed and incubated in 0.2 M HCl for 20 minutes, rinsed with water, digested with 0.5 mg/ml pronase in PBS at room temperature for 10 minutes, fixed in 4% paraformaldehyde for 20 minutes, rinsed with PBS, acetylated with 0.25% acetic anhydride/0.1 M triethanolamine for 10 minutes, rinsed with PBS, dehydrated through graded ethanol solutions, and air dried. A 25 μl aliquot of hybridisation mix was added to each section. The hybridisation mix consisted of 50% formamide, 2× saline sodium citrate (SSC), 10% dextran sulphate, 10 mM DTT, 250 μg/ml tRNA, and 1–4× 105 counts/min of radiolabelled RNA probe. The sections were covered with parafilm and incubated at 50°C overnight in an atmosphere of 50% formamide. After RNA–RNA hybridisation, the parafilm was removed and the sections were washed in a solution of 1× SSC, 50% formamide, and 10 mM DDT for four hours at 52°C, rinsed in 2× SSC for 20 minutes at 37°C, treated with 20 μg/ml of RNase A in 2× SSC for 30 minutes at 37°C, washed in 2× SSC and 0.1× SSC for 10 minutes each at room temperature, dehydrated through graded ethanols containing 0.3 M ammonium acetate, and air dried. Sections were then dipped into Ilford G5 emulsion, exposed at 4°C for three to 14 days, developed, fixed, and counterstained with haematoxylin and eosin.
The hybridisation mixture for in situ DNA–DNA hybridisation with the Cot1 probe was as for RNA–RNA in situ hybridisation. Instead of the radiolabelled RNA probes, the Cot1 probe was added at a 1/100 dilution of the labelling reaction mix. The sections were covered with parafilm and heated to 95°C for three minutes to denature target and probe DNA and then hybridised at 37°C overnight. After hybridisation, the sections were washed with 2× SSC for one hour at 37°C and then transferred to Tris buffered saline (TBS) for immunohistochemical detection of the bound probe with an antidigoxin monoclonal antibody (clone D1–22; Sigma, Poole, Dorset, UK) and the APAAP method. RNA–DNA in situ hybridisation using RNA probes generated from the pBSW plasmid was carried out as described for DNA–DNA in situ hybridisation. Double labelling combining immunohistochemistry for the detection of cellular antigens and in situ hybridisation for the detection of the EBERs, as well as double labelling immunohistochemistry for the simultaneous detection of the BZLF1 antigen and cellular antigens, were carried out as described previously.18
Results
EBV INFECTION IN A TONSIL FROM A PATIENT WITH INFECTIOUS MONONUCLEOSIS
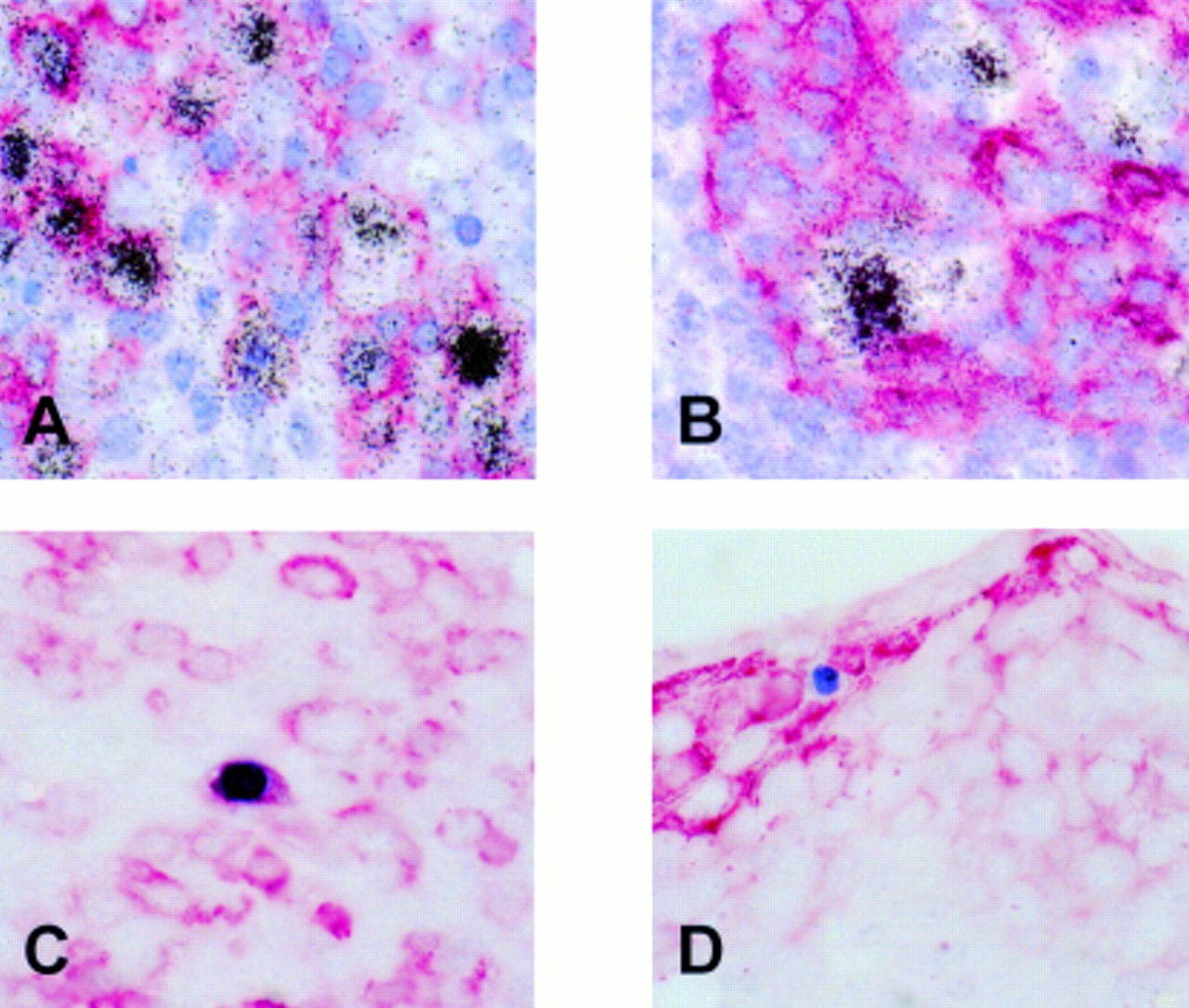
As reported previously, using EBER in situ hybridisation numerous EBV infected cells were identified in the tonsil, mainly in the extrafollicular areas.18 Double labelling revealed that most EBER expressing cells were CD20 positive B cells (fig 1A). EBER positive cells were seen frequently in the vicinity of tonsillar crypts. Double labelling showed that these cells were cytokeratin negative and thus were unlikely to be epithelial cells (fig 1B). Using immunohistochemistry, a small number of scattered BZLF1 staining cells were identified, indicating a switch from latent to replicative infection in a small subset of cells. Double labelling immunohistochemistry revealed the synthesis of BZLF1 in CD79a positive B cells showing morphological features of plasma cells (fig 1C) but not in cytokeratin positive epithelial cells (fig 1D).
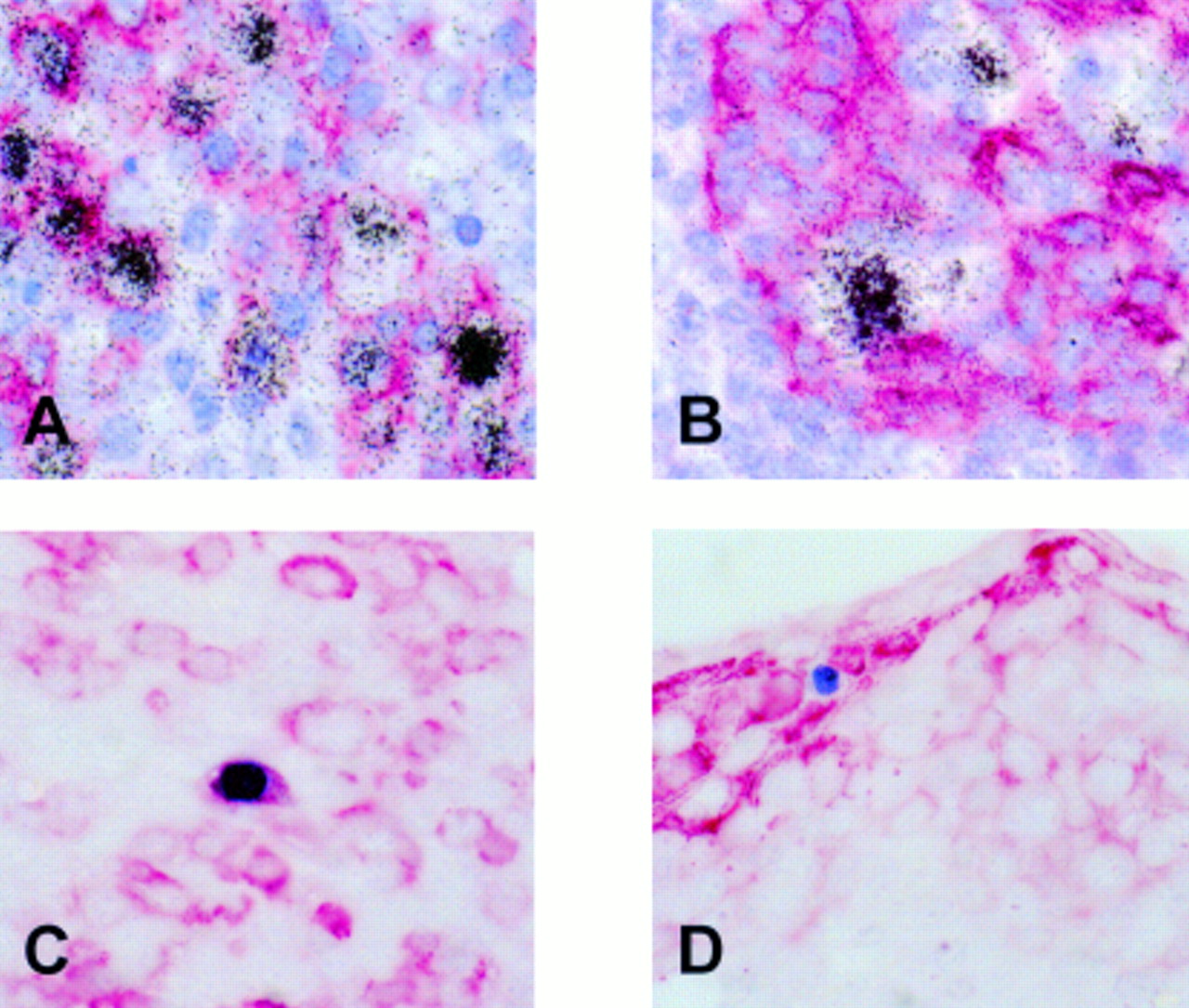
Detection of Epstein-Barr virus (EBV) infected cells in a tonsil from a patient with infectious mononucleosis. (A) Double labelling using radioactive in situ hybridisation (black silver grains) and immunohistochemistry (red staining) reveals numerous EBV encoded small RNA (EBER) positive/CD20 positive B cells. (B) These cells are often admixed with crypt epithelial cells but appear to be cytokeratin negative. (C) Production of the BZLF1 protein (blue staining) of EBV, indicating a switch from latent to replicative infection is seen in CD79a positive (red staining) B cells morphologically resembling plasma cells, but not in cytokeratin positive (red staining) epithelial cells (D).
VALIDATION OF THE CYTOBLOCK ANALYSIS
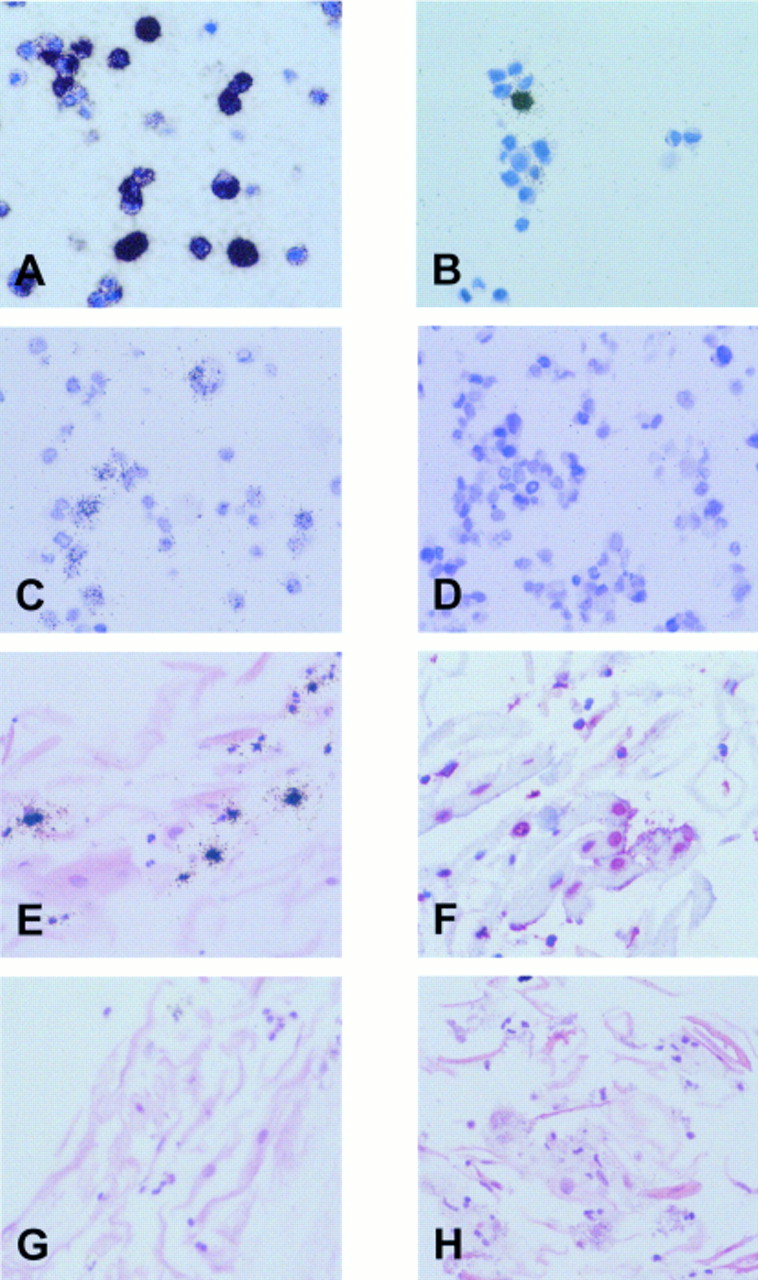
Using in situ hybridisation with EBER specific antisense probes, an intense signal was obtained in most B95.8 cells (fig 2A). A smaller proportion of cells was labelled using the sense control probes (fig 2B). These cells were similar in number to those staining for the BZLF1 protein (not shown), and this result probably reflects hybridisation of the sense RNA probe to replicating viral DNA present in a subset of cells, as reported previously.9 For the detection of EBV DNA, cellular DNA was denatured and in situ hybridisation was carried out with sense RNA probes derived from the pBSW plasmid. This procedure resulted in moderate labelling of a proportion of B95.8 cells, whereas no labelling was seen in the EBV negative Ramos cells (fig 2C and D).

{kind=link}
{kind=link}
Validation of cytoblock techniques. (A) In situ hybridisation with 35S labelled antisense probes reveals expression of Epstein-Barr virus (EBV) encoded small RNAs (EBERs) in most B95.8 cells. (B) A small proportion of B95.8 cells is also labelled with the sense control probes, indicating hybridisation to replicating viral DNA (see text). (C) In situ hybridisation with a probe specific for the BamHI W fragment of the EBV genome results in the labelling of a proportion of B95.8 cells, whereas the EBV negative Ramos cells (D) are unlabelled. (E) In situ hybridisation with a 35S labelled U6 specific RNA probe results in the accumulation of silver grains over scattered small inflammatory cells, whereas nuclei of epithelial cells are negative. (F) In situ hybridisation with a digoxigenin labelled Cot1 specific DNA probe results in red nuclear staining of most epithelial and inflammatory cells. (G) EBER specific in situ hybridisation with 35S labelled RNA probes shows no evidence of latent EBV infection in desquamated oropharyngeal epithelial or inflammatory cells from a patient with acute infectious mononucleosis. (H) Using a 35S labelled probe specific for the BamHI W fragment of the EBV genome, viral DNA is not detected in desquamated oropharyngeal cells.
To confirm that the cells present in the throat washings, particularly the densely keratinised desquamated epithelial cells, were accessible to the in situ hybridisation probes, all specimens were examined using a probe specific for cellular U6 RNA and a probe specific for Cot1 DNA. U6 is a ubiquitously expressed small RNA molecule that forms part of the spliceosome machinery. Like the EBERs, U6 is a nuclear RNA. In all cases, cells positive for U6 RNA were demonstrated, indicating RNA integrity (fig 2E). However, higher amounts of U6 were expressed in neutrophil polymorphonuclear cells and other inflammatory cells than in epithelial cells, which often appeared to be negative (fig 2E). This was investigated further using tissue sections from a palatinal tonsil. In tonsillar epithelium, U6 expression was weak or absent in terminally differentiated cells sloughing off at the surface of the squamous mucosa (not shown). This probably reflects a decrease in the metabolic activity of these terminally differentiated cells. Thus, the absence of detectable U6 expression in many desquamated epithelial cells in the throat washing samples appears to reflect a genuine lack of expression and does not seem to be a technical artefact.
The Cot1 DNA probe binds to Alu repeat sequences that are present at about 106 copies in the human genome. Binding of this probe was seen in the nuclei of epithelial cells and polymorphonuclear cells in the throat washings (fig 2F). This confirmed that the protocol used was suitable for the detection of nuclear DNA.
Sections from throat wash pellets were subjected to immunocytochemistry with a variety of antibodies to determine the nature of the cells present, but also to confirm that this material was suitable for immunocytochemical analysis. On morphological grounds, desquamated epithelial cells were the most prevalent cell type, with variable numbers of neutrophils and eosinophils admixed. This was confirmed by immunocytochemistry with anticytokeratin monoclonal antibodies, which resulted in strong cytoplasmic staining of the epithelial cells (not shown). The presence of numerous polymorphonuclear cells was confirmed by immunostaining with a CD15 monoclonal antibody (not shown). In contrast, B cells and T cells were only rarely detected, as shown by immunocytochemistry with CD20 and CD3 reagents, respectively (not shown). These results indicate that the formalin fixed and paraffin wax embedded throat washing cells were suitable for immunocytochemical analysis.
DETECTION OF EBV IN THROAT WASHINGS
Using EBER in situ hybridisation, labelled epithelial cells were not detected in any of the samples (fig 2G), indicating the absence of latent EBV infection from these cells. In contrast to a recent study, we did not see any EBV carrying B cells, probably because of the low number of B cells present in our samples (see above). Because the EBERs might be downregulated during the lytic cycle of EBV infection,19 we then searched for evidence of EBV replication. In situ hybridisation with a 35S labelled BamHI W DNA probe showed no specific labelling of epithelial cells. However, a small number of scattered inflammatory cells were labelled (not shown). A similar pattern was also seen with a vector control probe, and these cells were identified as eosinophils in haematoxylin and eosin stained sections. Thus, the signal seen in these cells with the BamHI W specific DNA probe reflects the non-specific binding of DNA probes to eosinophils, which has been reported previously.20 To circumvent this problem, we used RNA probes derived from the same plasmid in RNA–DNA hybridisation experiments. Using this approach, no labelling of any cells was seen in the throat washings (fig 2H). Thus, DNA in situ hybridisation indicated an absence of lytic EBV infection from desquamated epithelial cells. This conclusion was confirmed by immunocytochemistry for the detection of the BZLF1 transactivator protein of EBV, which also produced negative results in all cases (not shown).
Discussion
There is conflicting evidence with regard to the role of oropharyngeal epithelial cells in EBV infection. It has been proposed that primary and persistent EBV infection might be mediated through oropharyngeal epithelial cells.10 Support for this notion has come mainly from two sources. First, EBV replication has been detected in epithelial cells of oral hairy leukoplakia, an AIDS associated lesion of the tongue.8 Second, it has been reported that EBV infection is detectable in desquamated oropharyngeal epithelial cells from patients with infectious mononucleosis by in situ cytohybridisation.6,7 However, there is now increasing evidence pointing to B cells as the likely site of persistent EBV infection, and also as the possible target of primary EBV infection.22 Importantly, several studies have demonstrated that EBV replication in oral hairy leukoplakia is confined to the upper epithelial cell layers and is not accompanied by detectable latent infection of basal epithelial cells.9,14,15 Thus, these results do not support the idea that EBV persists in epithelial cells.
Here we confirm that, in tonsils from patients with infectious mononucleosis, EBV is detected mainly in B cells and not in epithelial cells, and that plasma cells can support EBV replication.16,18 However, in these tonsils EBV infected cells are often intimately admixed with crypt epithelial cells and thus it is difficult definitely to rule out EBV infection of occasional epithelial cells. Furthermore, it is conceivable that EBV might replicate in epithelial cells at other sites within the oropharynx. Therefore, we have re-examined the question of whether EBV infection is detectable in desquamated oropharyngeal epithelial cells from patients with acute infectious mononucleosis and from chronic virus carriers. To be able to apply a range of markers against latent and replicative EBV infection we decided to pellet the cells obtained by throat washing and to process them into paraffin wax embedded cytoblocks. This allowed us to cut multiple sections from different levels of the same specimen and to process sections for immunocytochemistry and in situ hybridisation as required. The analysis of similarly processed EBV positive (B95.8) and EBV negative (Ramos) cells confirmed that our methods were suitable for the detection of EBV DNA, the EBERs, and the BZLF1 protein in paraffin wax embedded cells. Furthermore, in situ hybridisation of sections from all throat wash samples with probes specific for Cot1 DNA and for U6 RNA produced the expected labelling patterns. Thus, these control experiments confirmed that the methods used were suitable for the detection of nucleic acids in the nuclei of the desquamated cells by in situ hybridisation.
We then attempted to detect markers of latent and lytic EBV infection in the throat washing cells. EBER specific in situ hybridisation is the most sensitive tool available for the detection of EBV latency. To detect lytic infection, immunocytochemistry with a BZLF1 specific monoclonal antibody and in situ hybridisation for the detection of the EBV genome were carried out. Using these methods, latent or lytic EBV infection was not detectable in epithelial cells in the throat washings from patients with infectious mononucleosis and from chronic virus carriers. This contrasts with studies by Sixbey et al and Lemon et al, but is in agreement with a more recent report.6,7,21 Karajannis et al have demonstrated the absence of EBV infection in desquamated oropharyngeal epithelial cells from patients with infectious mononucleosis.21 This observation is confirmed in our study. In addition, we have also demonstrated that there is no evidence of EBV infection in oropharyngeal epithelial cells from chronic virus carriers.
The reasons for the discrepant results of these and previous studies are uncertain. Because we have extensively validated our technical approach, we have no reason to assume that methodological problems have contributed to our failure to detect any evidence of EBV infection in desquamated oropharyngeal epithelial cells.
In summary, our data provide further evidence suggesting that epithelial cells are not a major site of EBV persistence and replication in vivo. These results are in keeping with the notion that B cells are the main site of EBV persistence and replication.22,23
Acknowledgments
This work was supported by grants from the Medical Research Council, from the United Birmingham Hospitals Endowment Fund, and from the Deutsche Forschungsgemeinschaft. We are most grateful to R Lisner and C Winkelmann for excellent technical assistance.