Article Text

Abstract
Aim—To characterise 12 Borrelia burgdorferi sensu lato isolates cultured from ticks collected in the Highlands of Scotland.
Methods—Three molecular methods were used: an outer surface membrane protein A (OSP A) gene polymerase chain reaction (PCR) designed to give different molecular weight products with different genomic groups, randomly amplified polymorphic DNA (RAPD) analysis, and ribosomal RNA (rRNA) gene PCRs using genomic group specific primers.
Results—All of the molecular methods used were quick and easy to perform and capable of differentiating between the different genomic groups of B burgdorferi sensu lato. All 12 tick isolates were characterised successfully with each method: five were characterised as B afzelii and seven were characterised as B burgdorferi sensu stricto. RAPD also identified differences within these genomic groups.
Conclusions—From this study, it is now known that at least two different B burgdorferi sensu lato genomic groups are present in the Highlands of Scotland: B afzelii and B burgdorferi sensu stricto. This information can now be used to develop appropriate serological tests, which should improve the diagnosis and management of patients with Lyme disease in Scotland. The molecular methods chosen were found to be useful typing tools and will allow rapid identification of any future isolates.
- Borrelia burgdorferi
- randomly amplified polymorphic DNA analysis
- ribosomal RNA gene polymerase chain reaction
Statistics from Altmetric.com
- Borrelia burgdorferi
- randomly amplified polymorphic DNA analysis
- ribosomal RNA gene polymerase chain reaction
Borrelia burgdorferi sensu lato is the spirochete responsible for the zoonotic infection, Lyme disease, but isolates are phenotypically and genotypically heterogeneous. They can be divided into at least 10 different genomic groups (or species) of B burgdorferi sensu lato: B afzelii, B garinii, B burgdorferi sensu stricto, B valaisiana, B lusitaniae, B andersonii, B japonica, B fanukii, B furdi, and B bissettii sp nov (formerly borrelia group DN127).1–3 These different genomic groups are associated with different geographical areas. Borrelia garinii, B afzelii, B valaisiana, B lusitaniae, and B burgdorferi sensu stricto are found in Europe; B burgdorferi sensu stricto, B andersonii, and B bissettii sp nov are found in North America; and B japonica, B fanukii, and B furdi are found in Japan. Only B garinii, B afzelii, and B burgdorferi sensu stricto are commonly isolated from human cases of Lyme disease, although in one study B bissettii sp nov was also isolated from humans.1,4
Although Lyme disease is endemic in many areas of the UK including the New Forest (Hampshire), Thetford Forest (Norfolk), Richmond Deer Park (London), Bushy Deer Park (London), throughout Ireland, Wales, and in the Scottish Highlands, it is not known which B burgdorferi sensu lato genomic groups are responsible.5–7 This is largely because of difficulties in growing and passaging the organism. Borrelia garinii has been successfully isolated in Ireland and B valaisiana has been detected in England.8,9 Recently, 12 B burgdorferi sensu lato isolates were successfully cultured from ticks collected in one location in the Highlands of Scotland.10 Our study set out to characterise the 12 Highland isolates using three molecular methods: an outer surface membrane protein A (OSP A) gene polymerase chain reaction (PCR) designed to give different molecular weight products with different genomic groups, randomly amplified polymorphic DNA (RAPD) analysis, and a PCR method using genomic group specific primers for the ribosomal RNA (rRNA) gene.11–13
Methods
CULTURE AND PREPARATION OF BORRELIA
Twelve isolates of B burgdorferi were cultured from ticks collected on three separate occasions in the summer of 1997 from two locations approximately five and 10 miles to the east of Inverness in the Highlands of Scotland.10 Both locations were wooded and the ticks were collected by sheet dragging the grassy areas under the trees. A total of 128 ticks were collected, giving an overall positivity rate of 9.4%.
Four reference strains (B afzelii ACA-1, B garinii A57, B burgdorferi sensu stricto Hb1, and B garinii A60) and the 12 isolates (C7, E5, E8, E9, E10, F2, F8, G4, G5, G10, H1, and J1) of B burgdorferi were cultured in Barbour-Stoenner-Kelly (BSK)-H medium (Sigma-Aldrich, Poole, Dorset, UK) supplemented with 6.6% inactivated rabbit serum (Sigma-Aldrich), 1.4% gelatin (Sigma-Aldrich), 0.8% septrin (GlaxoWellcome, Uxbridge, Greater London, UK; trimethoprim 16 mg/litre, sulphamethoxazole 80 mg/litre) and 0.8% neomycin (GlaxoWellcome; 100 mg/litre). Cultures were incubated at 33°C under microaerophilic conditions.10 The organisms were harvested by centrifugation at 10 000 ×g for 30 minutes and the pellets washed twice in 0.9% (wt/vol) sodium chloride (Steripak, Runcorn, Cheshire, UK). They were then centrifuged again at 10 000 ×g for 30 minutes and resuspended in minimal amounts of sterile distilled water. The DNA was extracted with proteinase K (Sigma-Aldrich; 200 mg/litre) in Tris EDTA buffer containing 0.5% Tween 20 and then quantified by spectrophotometry (260 nm).
OSP A GENE PCR
The primary OSP A gene PCR, described previously by Guttman et al,11 was modified (number of cycles increased) to produce three different sized products of approximately 704 bp, 980 bp, and 1107 bp.11 A 50 μl reaction mixture was used for each sample and contained 105 organisms, 10 mM Tris/HCl (pH 8.5), 50 mM KCl, 2.5 mM MgCl2 (Advanced Biotechnologies, Epsom, Surrey, UK), 0.2 mM dNTPs (Pharmacia Biosystems, Milton Keynes, Buckinghamshire, UK), 0.5 μM of primer 1 (OSP1: 5′-AAA AAA TAT TTA TTG GGA ATA GG-3′), 0.5 μM of primer 2 (OSP2: 5′-GTT TTT TTG CTG TTT ACA CTA ATT GTT AA-3′; Severn Biotech, Kidderminster, UK) and 0.5 U Taq DNA polymerase (Advanced Biotechnologies). After an initial denaturation step of 96°C for two minutes, 40 cycles of 30 seconds at 94°C, 30 seconds at 37°C, and two minutes at 72°C were carried out, followed by a final extension period of five minutes at 72°C. The products were then separated by electrophoresis using a 10% polyacrylamide gel run in Tris borate EDTA (TBE) buffer at 1.5 V for four hours. After staining the gel in TBE buffer containing 1 mg/litre ethidium bromide, the bands were visualised with a UV transilluminator.
RAPD-PCR ASSAY
A previously described RAPD-PCR assay was optimised.1 During optimisation each component of the reaction mixture was evaluated: seven different primers were tried (BB1: 5′-CCG CAG CCA A-3′, BB2: 5′-GCG ATC CCC A-3′, BB3: 5′-AAG AGC CCG T-3′, F1: 5′-ATT AAC GCT GCT AAT CTT AGT-3′, F2: 5′-GTA CTA TTC TTT ATA GAT TC-3′, OSP1, and OSP2—described above); 0.1 mg/ml or no bovine serum albumin (BSA; Sigma-Aldrich); different MgCl2 concentrations (1–4 mM); different DNA concentrations; different polymerase enzyme concentrations (1–4 U); and five different types of polymerase enzymes (Taq DNA polymerase, Advanced Biotechnologies; Thermostable DNA polymerase, Advanced Biotechnologies; Extensor Long PCR system, Advanced Biotechnologies; Taq DNA polymerase Gold, Bio/Gene, Cambridge, UK; and Taq Supreme, Helena Biosciences, Sunderland, Tyne and Wear, UK).
The optimised RAPD-PCR was performed with a 50 μl reaction mix containing 40 ng DNA (2.5 × 107 organisms),14 10 mM Tris/HCl (pH 8.5), 50 mM KCl, 4 mM MgCl2, 0.8 mM dNTPs, 0.04 mM of one primer (BB1 or OSP1), no BSA, and 3.5 U of the Taq DNA polymerase (Advanced Biotechnologies). After an initial denaturation step of two minutes at 94°C, we carried out three cycles of: five minutes at 94°C, five minutes at 36°C, and five minutes at 72°C, followed by 40 cycles of: one minute at 94°C, one minute at 36°C, and two minutes at 72°C. A final extension step of 10 minutes at 72°C then completed the RAPD-PCR. RAPD-PCR products were electrophoresed on 1% agarose 20 × 20 cm gels containing 1 mg/litre ethidium bromide in TBE buffer at 133 V for two hours, or electrophoresed on 15 × 11 cm agarose gels for two hours, stained for 30 minutes to one hour in TBE buffer containing 1 mg/litre ethidium bromide, and then visualised using a UV transilluminator. A MultiVariate Statistics Package (MVSP), version 2.1c was used to analyse the resulting RAPD fingerprints.
SPECIFIC rRNA GENE PCRS
Specific rRNA gene PCR primers previously described by Liebisch et al for B afzelii (BA1: 5′-GCA TGC AAG TCA AAC GGA-3′ and BA2: 5′-ATA TAG TTT CCA ACA TAG C-3′) and B burgdorferi sensu stricto (BBS1: 5′-GGG ATG TAG CAA TAC ATT C-3′ and BBS2: 5′-ATA TAG TTT CCA ACA TAG G-3′) were used to amplify products of 589 bp and 574 bp, respectively.12 The PCRs were performed with a 50 μl reaction mix containing 1.6 pg DNA (103 organisms), 10 mM Tris/HCl (pH 8.5), 50 mM KCl, 2.5 mM MgCl2, 0.2 mM dNTP, 1 μM of each primer, and 0.5 U Taq DNA polymerase. All reaction mixes were subjected to 40 cycles of 94°C for 90 seconds, 42°C or 48°C, respectively, for two minutes, and 72°C for two minutes, followed by a further extension period of 72°C for five minutes. The PCR products were electrophoresed on 2.5% 15 × 11 cm agarose gels containing 1 mg/litre ethidium bromide in TBE buffer at 133 V for 20 minutes and then visualised using a UV transilluminator.
Results
OSP A GENE PCR
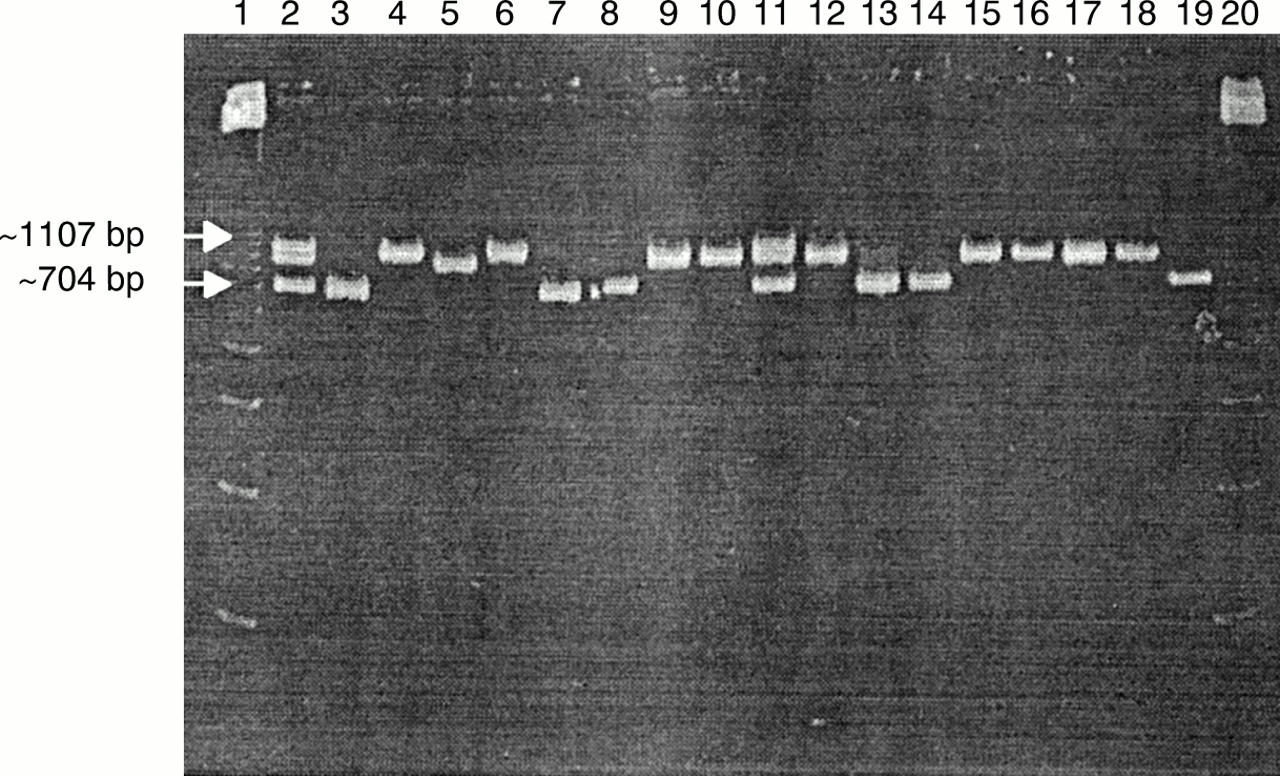
The four reference strains—B afzelii ACA-1, B garinii A57, B burgdorferi sensu stricto Hb1, and B garinii A60—and 12 isolates all produced OSP A gene PCR products. Reference strain B afzelii and tick isolates C7, E5, F2, F8, and J1 gave rise to products with the lowest molecular weight (∼ 704 bp); reference strain B burgdorferi sensu stricto and tick isolates E8, E9, E10, G4, G5, G10, and H1 gave rise to products with an intermediate molecular weight (∼ 980 bp), but only reference strains B garinii A57 and A60 gave rise to products with the highest molecular weight (∼ 1107 bp) (fig 1).

Polyacrylamide gel electrophoresis of outer surface membrane protein A (OSP A) gene specific polymerase chain reaction (PCR) products illustrating three different molecular weights: approximately 704 bp from reference strain Borrelia afzelii ACA-1 (lane 3) and tick isolates C7, E5, F2, F8, and J1 (lanes 7, 8, 13, 14, and 19, respectively); approximately 1107 bp from reference strains B garinii A57 (lane 4) and B garinii A60 (lane 6); approximately 980 bp from reference strain B burgdorferi sensu stricto Hb1 (lane 5) and tick isolates E8, E9, E10, G4, G5, G10, and H1 (lanes 9, 10, 12, 15, 16, 17, and 18, respectively); and all three bands from mixes of reference strains B afzelii ACA-1, B garinii A57, and B burgdorferi sensu stricto Hb1 (lanes 2 and 11). Lanes 1 and 20 contain a 123 bp ladder.
RAPD ASSAY
During the optimisation of the RAPD assay, five of the seven primers tried gave results: BB1, BB2, BB3, OSP1, and OSP2. Of these primers, BB1 and OSP1 gave the best differentiation between the reference strains. BSA was not found to be useful and the MgCl2 concentration could not be reduced below 4 mM. Preliminary results showed that DNA concentrations of 0.2 ng produced too few bands compared with 20 ng DNA, and enzyme concentrations of 3–4 U were required to produce sufficient bands. For subsequent experiments, 3.5 U of enzyme were used. Similar results were achieved with 5–160 ng DNA. Four of the polymerase enzymes tried produced similar, but not identical, fingerprints: Taq, Thermostable, Taq Gold, and Extensor mix (fig 2). Taq produced the best differentiation of B afzelii and B burgdorferi sensu stricto products, and this enzyme was used in subsequent experiments. Taq Supreme produced very different fingerprints, with the presence of many lower molecular weight bands.

Agarose gel electrophoresis illustrating similar, but not identical randomly amplified polymorphic DNA (RAPD) fingerprints with four different polymerase enzymes: Taq (lanes 2 and 3), Thermostable (lanes 4 and 5), Taq Gold (lanes 6 and 7), and Extensor mix (lanes 8 and 9). Also included were: 40 ng Borrelia afzelii ACA-1 (lanes 2, 4, 6, and 8) or B burgdorferi sensu stricto Hb1 (lanes 3, 5, 7, and 9), primer BB1, 3.5 U enzyme, and 4 mM MgCl2. Lane 1 contains a 123 bp ladder.
Using the optimised RAPD assay (4 mM MgCl2, 3.5 U Taq, and 40 ng DNA) with primers BB1 and OSP1 all of the reference strains and tick isolates produced good fingerprints containing five to 10 bands. Three overall fingerprint patterns were seen for each primer, classified as B1, B2, and B3 with primer BB1; and O1, O2, and O3 with primer OSP1. Reference strain B afzelii ACA-1 and tick isolates C7, E5, F2, F8, and J1 produced B1 and O1 fingerprints; reference strains B garinii A57 and A60 produced B2 and O2 fingerprints; and reference strain B burgdorferi sensu stricto and tick isolates E8, E9, E10, G4, G5, G10, and H1 produced B3 and O3 fingerprints (figs 3 and 4). Within these overall patterns small differences were visible. These differences were highlighted in the dendrogram produced using the MVSP analysis package (fig 5). All of the tick isolates differed from the reference strains. Five (E5, F2, F8, C7, and J1) of the isolates grouped with B afzelii ACA-1 and seven (E8, G10, H1, E9, G5, G4, and E10) grouped with B burgdorferi sensu stricto Hb1. Differences within both B afzelii and B burgdorferi sensu stricto groups were found, with E10 being the most divergent.
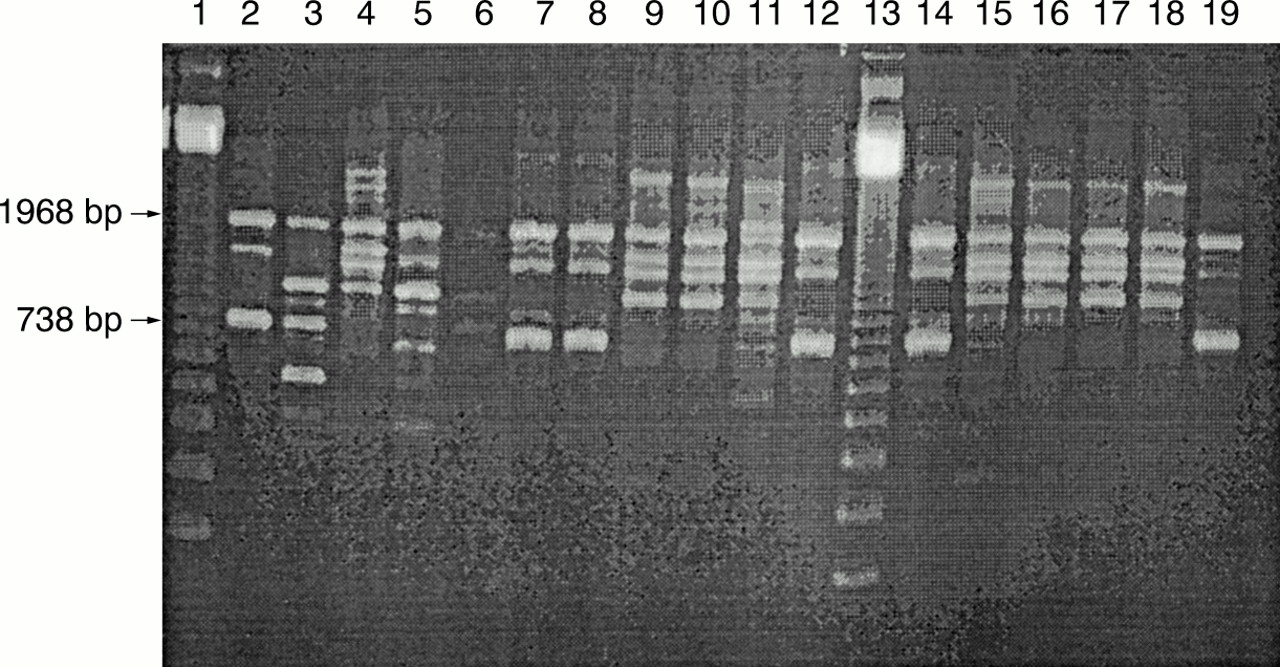
Agarose gel electrophoresis illustrating randomly amplified polymorphic DNA (RAPD) fingerprints of the four reference strains and 12 tick isolates produced with primer BB1, 3.5 U Taq DNA polymerase, 4 mM MgCl2, and 40 ng DNA. Three main RAPD fingerprints were obtained: B1 from reference strain Borrelia afzelii ACA-1 (lane 2) and tick isolates C7, E5, F2, F8, and J1 (lanes 7, 8, 12, 14, and 19, respectively); B2 from reference strains B garinii A57 (lane 3) and B garinii A60 (lane 5); and B3 from reference strain B burgdorferi sensu stricto Hb1 (lane 4) and tick isolates E8, E9, E10, G4, G5, G10, and H1 (lanes 9, 10, 11, 15, 16, 17, and 18, respectively). Lane 6 contains negative control (distilled water) and lanes 1 and 13 contain a 123 bp ladder.
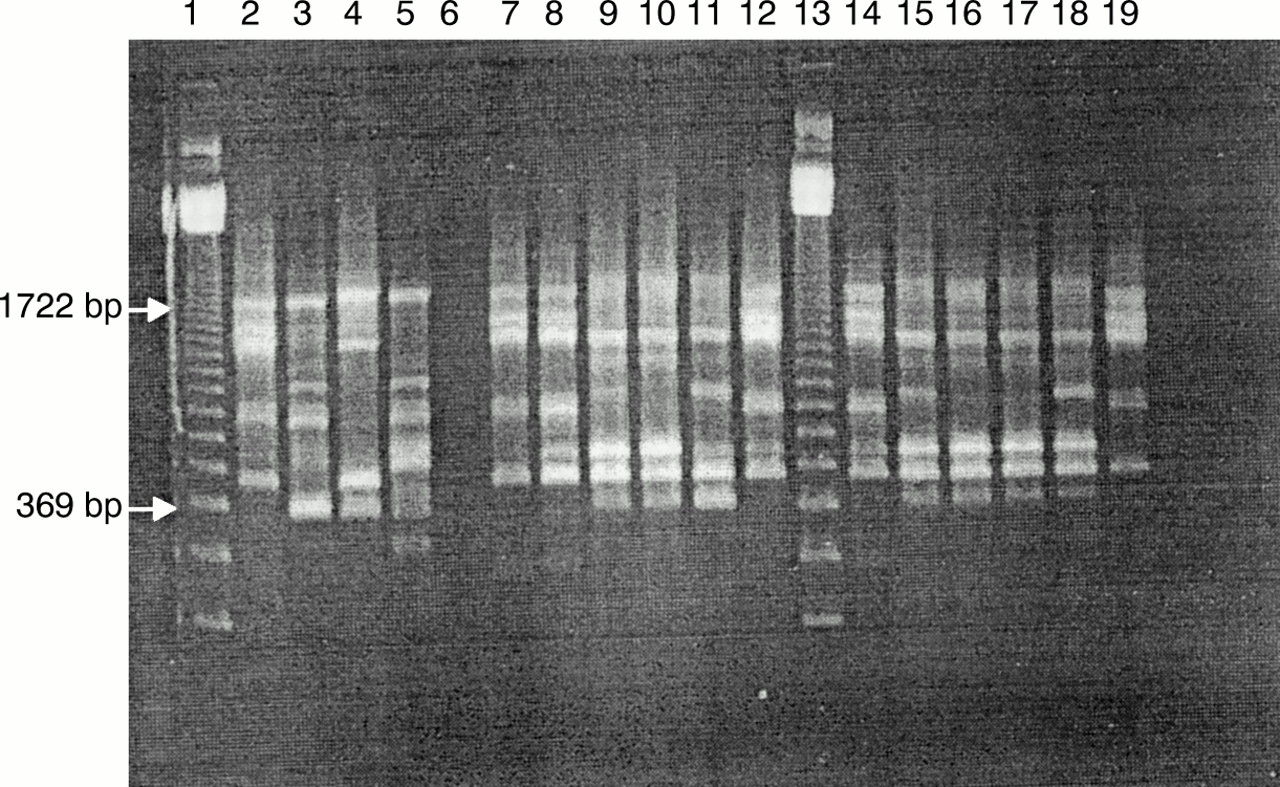
Agarose gel electrophoresis illustrating randomly amplified polymorphic DNA (RAPD) fingerprints of the four reference strains and 12 tick isolates produced with primer OSP1. Three main RAPD fingerprints were obtained: O1 from reference strain Borrelia afzelii ACA-1 (lane 2) and tick isolates C7, E5, F2, F8, and J1 (lanes 7, 8, 12, 14, and 19, respectively); O2 from reference strains B garinii A57 (lane 3) and B garinii A60 (lane 5); and O3 from reference strain B burgdorferi sensu stricto Hb1 (lane 4) and tick isolates E8, E9, E10, G4, G5, G10, and H1 (lanes 9, 10, 11, 15, 16, 17, and 18, respectively) with 3.5 U Taq DNA polymerase, 4 mM MgCl2, and 40 ng DNA. Lane 6 contains negative control and lanes 1 and 13 contain a 123 bp ladder.

Dendrogram of combined primer BB1 and OSP1 randomly amplified polymorphic DNA (RAPD) results illustrating that tick isolates E5, F2, F8, C7, and J1 are most similar to reference strain Borrelia afzelii ACA-1 and tick isolates E8, G10, H1, E9, G5, G4, and E10 are most similar to reference strain B burgdorferi sensu stricto Hb1. The B afzelii tick isolates are of two types (E5, F2, F8 or C7, J1) and B burgdorferi sensu stricto tick isolates are of four types (E8, G10, H1; or E9, G5; or G4; or E10).
SPECIFIC rRNA GENE PCRS
When using the genomic group specific primers for B afzelii and B burgdorferi sensu stricto, reference strain B afzelii ACA-1 and tick isolates C7, E5, F2, F8, and J1 were positive only with B afzelii specific primers; reference strain B burgdorferi sensu stricto Hb1 and tick isolates E8, E9, E10, G4, G5, G10, and H1 were positive only with B burgdorferi sensu stricto specific primers; and reference strains B garinii A57 and A60 were negative with both sets of primers (fig 6).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Agarose gel electrophoresis of ribosomal RNA gene polymerase chain reaction (PCR) products using Borrelia afzelii (top) and B burgdorferi sensu stricto (bottom) specific primers. Reference strain B afzelii ACA-1 (lane 2) and tick isolates C7, E5, F2, F8, and J1 (lanes 6, 7, 11, 12, and 17, respectively) were B afzelii positive; reference strains B garinii A57 (lane 3) and B garinii A60 (lane 5) were negative; and reference strain B burgdorferi sensu stricto HB1 (lane 4) and tick isolates E8, E9, E10, G4, G5, G10, and H1 (lanes 8, 9, 10, 13,14 15, and 16, respectively) were B burgdorferi sensu stricto positive. Lanes 1 and 18 contain a 123 bp ladder.
Discussion
Numerous methods have been used to type B burgdorferi sensu lato isolates including ribotyping, DNA–DNA hybridisation, 16S rRNA sequencing, plasmid profiling, and restriction fragment length polymorphism analysis.14 The three molecular methods used in our study were chosen because they required little specialised equipment, were relatively inexpensive, and were easy to perform. The OSP A gene and rRNA gene PCRs were used so that the genomic group of the isolates could be clearly defined. The RAPD assay was used for the same purpose, and also to differentiate any differences between strains within these genomic groups. Although the three methods were easy to perform, the RAPD assay described by Wang et al had to be optimised for use in our laboratory.1 It was found that magnesium chloride concentration and enzyme concentration, source, and type all had crucial effects on the RAPD fingerprints produced. However, very similar RAPD fingerprints were obtained within a wide range of DNA concentrations, 5–160 ng, and we chose 40 ng. Once the assay had been fully optimised, reproducible results were obtained. Because of the differences in product (sometimes small but consistent) produced by different polymerases, it appears to be important to choose a suitable enzyme and use it for all analyses. The polymerase in our study was chosen because it gave the most clear cut differentiation of strains with both primers used.
The three methods clearly differentiated the reference strains and each was able to characterise the Highland tick isolates into a specific genomic group. The results of the three different methods were in full agreement, with five of the tick isolates being characterised as B afzelii strains and seven as B burgdorferi sensu stricto strains. In addition, the RAPD analysis was able to distinguish differences within all three genomic groups investigated. Two different Scottish B afzelii strains were identified and four different B burgdorferi sensu stricto strains. These all differed from the reference strains. The presence of different strains in Scotland compared with other areas of Europe, and different strains within one small area of Scotland, is not surprising because many previous studies have also identified strain heterogeneity within these genomic groups.1,15
A large European study detected the simultaneous presence of several genomic groups in a high proportion of infected ticks and concluded that this is probably the rule and not an exception in European Lyme disease.12 No mixed infections were detected in our study; however, because all tests were carried out on cultured isolates, not tick extracts, there may have been selection of certain genomic groups during cultivation.16 In other studies, multiple genotypes were found when samples were directly investigated but they were not found in culture samples.17,18
Previous studies have only detected B garinii and B valaisiana in the UK.8,9 Therefore, it is surprising that neither of these genomic groups was found during our study. Although B afzelii and B burgdorferi sensu stricto might be the only genomic groups in the small geographical area studied, other genomic groups might be present in other parts of Scotland. Other studies have found pronounced differences in the prevalence of different genomic groups depending on the area sampled. For example, in Finland B burgdorferi sensu stricto has only been detected in the southwestern parts of the country, and the prevalence of B garinii seems to increase towards the eastern borders. One suggested explanation for this is that the different genomic groups exist in different animal reservoirs.18
The finding of two different genomic groups in Scotland and the identification of different strains within these genomic groups has important implications for diagnosis. Currently, laboratory diagnosis relies heavily on serological tests. However, it is recognised that test sensitivity and specificity vary considerably depending on the antigens used. Recently, improved tests have been developed using local antigens representing the most common genomic groups present in the test area.19,20 This is because different genomic groups and different strains within the genomic groups produce different immune responses.21 In our laboratory, a commercial enzyme linked immunosorbent assay (ELISA) is used for screening and in house immunoblotting for confirmation. The antigens for these tests are derived from American and European reference strains of B burgdorferi sensu stricto. An in house ELISA is also available using antigen from the same reference strain as is used for immunoblotting.10 Although B burgdorferi sensu stricto was found in our study, the RAPD patterns were not identical to those of the European strain used in our in house ELISA and immunoblotting techniques. Therefore, local strains of B burgdorferi sensu stricto and B afzelii could improve the sensitivity and specificity of our in house serological tests.
Acknowledgments
We thank Dr J Ball (QMC Nottingham) for his help and support during the optimisation of the RAPD test. We would also like to thank Dr J Robertson and Dr S O'Connell (Southampton PHLS) for generously supplying reference strains Borrelia afzelii ACA-1, B garinii A57, B burgdorferi sensu stricto, and B garinii A60; Dr R Evans (Raigmore Hospital) for the initial introduction of the OSP A PCR into our laboratory and helpful discussions; and Dr A Deacon (Raigmore Hospital) and Dr D Morrison (MRSA reference laboratory, Glasgow) for their assistance during computer analysis of the results. We would also like to thank Ms M Forbes for secretarial assistance.