Article Text

Abstract
Hodgkin's disease is an unusual cancer because the malignant cells constitute only a minority of the total tumour mass and, as a consequence, the study of these cells has been a major challenge. Recently, the application of newer technologies, such as single cell polymerase chain reaction (PCR) and gene expression array analysis, to the study of Hodgkin's disease have yielded new insights into the pathogenesis of this tumour. In addition, the recognition that a proportion of Hodgkin's disease tumours harbour the Epstein-Barr virus (EBV) and that its genome is monoclonal in these tumours suggests that the virus contributes to the development of Hodgkin's disease in some cases. This review summarises current knowledge of the pathogenesis of Hodgkin's disease with particular emphasis on the association with EBV. J Clin Pathol: Mol Pathol
- Hodgkin's disease
- Epstein-Barr virus
- Hodgkin-Reed Sternberg cells
Statistics from Altmetric.com
The histological expression of Hodgkin's disease is diverse. The varied patterns in cellular make up and nodal architecture have caused difficulty in the diagnosis and classification of the disease, and there have been numerous histological classification systems. The first was proposed by Rosenthal in 1936,1 and was based on the observation that prognosis for patients with Hodgkin's disease is related to the number of lymphocytes in tissue sections of their tumours. Other systems proposed after Rosenthal's initial work included Jackson and Parker's in 19442 and Lukes and Butler's in 1966,3 the latter being modified at the Rye conference.4 The Rye modification of the Lukes and Butler classification divides Hodgkin's disease into four histological subtypes: lymphocyte predominance (LP), nodular sclerosis (NS), mixed cellularity (MC), and lymphocyte depletion (LD). The more recent revised European American lymphoma (REAL) classification5 identifies LP, NS, MC, LD, and a provisional entity known as lymphocyte rich classic Hodgkin's disease.
The diagnostic lesion of Hodgkin's disease is characterised by the disruption of normal lymph node architecture and the presence of a minority, usually less than 1–2% of the total tumour mass, of malignant Hodgkin-Reed Sternberg (HRS) cells amid a background of non-neoplastic cell populations.5,6 The typical cellular background comprises T and B cells, eosinophils, neutrophils, plasma cells, histiocytes, fibroblasts, and stromal cells, which either surround the HRS cells or accumulate in their close vicinity.5 The HRS cells and their reactive neighbouring cells are able to crosstalk via a complex of cytokine and cell contact dependent interactions, and these probably include proliferative and anti-apoptotic signals favouring tumour cell survival and expansion.7 The diagnostic separation of the histological subtypes relies on the type and relative proportions of these so called reactive bystander cells, the presence of fibrosis, and the detection of the various morphological variants of HRS cells.5
Immunohistological studies and clinical investigations performed in the 1980s revealed that the subtypes NS, MC, and LD are immunophenotypically and clinically similar to each other but different from the LP subtype.8,9 This finding is taken into account in the REAL classification,5 which categorises the subtypes NS, MC, and LD under the term “classic Hodgkin's disease”. The LP subtype is now considered to be a distinct entity because diagnostic HRS cells expressing CD15 or CD30 are rarely encountered,10 whereas B cell markers such as CD20 are regularly expressed by tumour cells.5
Origin of the malignant cells in Hodgkin's disease
The relative scarcity of HRS cells within Hodgkin's disease tumours has made their study problematic. Early phenotypic studies suggested lineages related to macrophages or histiocytes,11 dendritic cells,12 or granulocytes.13 However, evidence now supports the idea that the HRS cell originates from either a B or T cell.14–18
Many recent studies have relied on the use of polymerase chain reaction (PCR) analysis of single HRS cells to provide information on their clonality and potential cell of origin.19 Studies investigating the presence of rearranged VH genes in individual HRS cells have produced conflicting results. One group detected polyclonality in four patients with LP Hodgkin's disease,20 whereas another found monoclonality in patients with the LP, NS, and MC forms of Hodgkin's disease (one patient each).19 A larger series demonstrated a range of clonality patterns, including monoclonal B cells in three of eight cases of MC Hodgkin's disease, and a mixed monoclonal and polyclonal pattern in an additional three cases.21 The remaining patients, including all four with NS, had polyclonal patterns. Similar results were reported by Delabie et al,22 who found some cases of NS Hodgkin's disease with a B cell phenotype and polyclonal immunoglobulin heavy chain (IgH) rearrangements. These findings suggested that the evolution of classic Hodgkin's disease might involve the progression from polyclonal to monoclonal disease. A study of relapsed patients with classic Hodgkin's disease showed that the same IgH rearrangements with identical somatic mutations were found from biopsies taken over a period of three years, demonstrating the persistence and dissemination of a clonal tumour cell population.23 Further analysis has revealed that HRS cells carry high loads of somatic Ig mutations, indicating that they originate from germinal centre or post-germinal centre B cells. In most cases it was shown that the VH gene rearrangements were non-functional, suggesting that HRS cells can bypass apoptosis, which would otherwise eliminate B cells with defectively rearranged Ig genes.24
However, using a single cell PCR approach with less risk of contamination, Marafioti et al only detected monoclonal Ig rearrangements in HRS cells, most of which did not disrupt the Ig coding capacity.25 This suggests that some of the previous findings of polyclonal populations of HRS cells might have been the result of technical artefacts. In this study, the absence of Ig expression in HRS cells was confirmed by in situ hybridisation, but clearly could not be explained by the presence of crippling mutations.
The absence of Ig gene expression in classic Hodgkin's disease contrasts with results from the analysis of LP Hodgkin's disease, where the tumour cell population often expresses Ig.9 Furthermore, there are differences in the nature of IgH mutations between classic and LP Hodgkin's disease. Whereas intraclonal diversity is uncommon in classic Hodgkin's disease it is more frequent in LP Hodgkin's disease, suggesting the presence of ongoing somatic mutations in the LP form but not the classic form of the disease.26
Occasionally, Hodgkin's disease tumours express T cell antigens, including granzyme B and T cell intracellular antigen 1 (TIA-1). In some cases, this has been shown to represent aberrant expression of T cell antigens by HRS cells that show evidence of IgH gene rearrangement and are thus assumed to be B cell in origin.27 However, in the same study a single Hodgkin's disease case showed expression of T cell markers and also T cell receptor (TCR) gene rearrangements, indicating that at least a minority of HRS cells are genuinely of T cell origin.
Newer technologies have provided the means of global gene expression analysis in HRS cells. Cossman et al used cDNA libraries prepared from single HRS cells of primary tissues to analyse gene expression and compare this to similar libraries derived from germinal centre B cells and dendritic cells.28 This study provided further support for a B cell origin for HRS cells, based on the frequent detection of markers such as BL34 and B7.1-CD80, and was also able to identify genes such as the melanoma associated tumour antigen, MAGE-4a, and the transcription factor, Pax-6, not previously known to be expressed in Hodgkin's disease. In the second of such studies, microarray analysis identified the interleukin 13 (IL-13) gene to be highly expressed in Hodgkin's disease derived cell lines.29 Subsequent in situ hybridisation of lymph node tissue from patients with Hodgkin's disease showed that HRS cells specifically expressed high amounts of IL-13. Although such techniques are powerful and provide the means to identify novel genes expressed in HRS cells, unravelling the role of these genes and their relevance to the biology of Hodgkin's disease will need careful investigation.
Epidemiology of Hodgkin's disease
In 1966, Brian MacMahon,30 reviewing the epidemiology of Hodgkin's disease, identified a bimodal age distribution in the USA, with the first peak of clinical onset occurring between 15 and 34 years, and the second after 50 years of age. Three age periods were distinguished: 0–14, 15–34, and 50 years and above. MacMahon noted that childhood cases of Hodgkin's disease were more common in boys (85% boys, 15% girls) for children less than 10 years of age. In young adults he hypothesised that the disease was probably infectious in nature, with low infectivity. This was supported by data from some families that had more than one affected member of different ages at the same time31,32; an excess of cases had also been identified in winter months.33,34 The peak incidence in young adults was between 25 and 30 years of age, the sex ratio was almost equal at this time, and the disease was associated with high socioeconomic status as defined by the Registrar General.30 MacMahon also identified that the disease in the elderly showed increasing incidence with age, a male to female patient ratio of 2 : 1, and epidemiological features similar to other neoplastic diseases, such as chronic lymphatic leukaemia. MacMahon30 hypothesised that this pattern was typical of neoplastic disease and was quite distinct from the young adult disease.
Correa and O'Conor35 introduced the concept of at least three epidemiological patterns of Hodgkin's disease based upon country of residence. A type I pattern is characterised by relatively high incidence rates in male children, low incidence in the third decade, and a second peak of high incidence in older age groups. The histological subtypes are often those with a less favourable prognosis, usually either MC or LD. This pattern prevails in developing countries.
Type III is the converse of the type I pattern, being characterised by low rates in children and a pronounced initial peak in young adults. The more favourable subtype of NS is common and this pattern is typical of developed countries.
Type II is an intermediate pattern found—for example, in rural areas of developed countries, and reflects a transition between type I and type III. Correa and O'Conor35 interpreted these data as the result of the interplay of environmental and host factors influencing the natural history of a single disease, and likened it to tuberculosis. In underprivileged communities, there are higher rates of tuberculosis in children and the disease presents itself in the more serious pneumonic form. When economic conditions improve, childhood tuberculosis becomes less common and most cases in young adults are of the more benign pulmonary form. This led to the hypothesis that in a given population susceptibility to the agent or agents that cause Hodgkin's disease is related to immunocompetence and host response, the degree of which is, in turn, dependent on environmental and socioeconomic factors. Hence, there is an alternative to the dual aetiology explanation of bimodality; that of a single aetiological process that is affected by variations in host response over age.
Childhood social environment has been suggested to play an important role in influencing the risk of Hodgkin's disease among young adults,36 with higher risk being associated with factors that diminish or delay the exposure to infectious agents, such as higher social class, more education, small family size, and early birth order position. These are consistent with a virus induced pathogenesis, with greater risk of Hodgkin's disease occurring with increasing age at infection.
Gutensohn and Cole37 likened the epidemiology of Hodgkin's disease in young adults to that of paralytic poliomyelitis in the pre-vaccine era. In both diseases, age of peak incidence is delayed as living conditions improve. For both, increased risk is associated with higher social class and small family size. They suggested that Hodgkin's disease is a rare consequence of a common infection, with the probability of the disease increasing as age at the time of infection increases.
These findings led to the premise that the variation of the bimodal age incidence curve of Hodgkin's disease is related to the age at primary infection with a common virus. As a population moved towards a higher standard of living, an initial early peak among young boys disappeared and produced the characteristic young adult peak.36 The data from the study of factors in childhood environment that influence the age of infection are consistent with this idea. The incidence of disease in the older age group varies little between populations and is not associated with social class factors.38
Epstein-Barr virus and Hodgkin's disease
As early as 1966 MacMahon30 proposed that Hodgkin's disease might be caused by an infectious agent. The first evidence that this agent might be Epstein-Barr virus (EBV) was provided by the detection of raised antibody titres to EBV antigens in patients with Hodgkin's disease when compared with patients with other lymphomas39 and, further, that these raised values preceded the development of Hodgkin's disease by several years.40 In addition, the relative risk of developing Hodgkin's disease in individuals with a history of infectious mononucleosis, relative to those with no previous history, was shown to range between 2.0 and 5.0.36 However, antibody titres to other herpesviruses, including human herpesvirus 6, have been shown to be raised in prediagnostic sera from patients with Hodgkin's disease,41 although these antibody titres were higher in EBV negative as opposed to EBV positive cases.42 In addition, raised antibody titres to the EBV viral capsid antigen do not predict EBV status in Hodgkin's disease.43
EBV could either play a direct or indirect role in the pathogenesis of Hodgkin's disease, possibly by triggering the pathogenic mechanism(s), or it could reflect the presence of an inherited or acquired depression of immunoregulation that is a prelude both to the malignancy and to the reactivation of EBV.44 Immunosuppressed patients show rises in all herpesvirus antibodies, rather than a selective rise in EBV antibodies,45 which suggests that depression of immunoregulation, rather than a specific disease phenomenon, might be responsible for these raised values.
With the advent of cloned viral probes and Southern blot hybridisation methods, EBV DNA was initially detected in 20–25% of Hodgkin's disease tumour specimens.46 However, this approach could not determine the locality of the EBV genome in tissues. In situ hybridisation methods to detect EBV DNA provided the first demonstration of its existence in the HRS cells.47–49 Subsequently, the demonstration of the abundant EBV early RNA (EBER1 and EBER2) sequences in HRS cells provided a sensitive method for detecting latent infection in situ. This technique is generally accepted as the “gold standard” for the detection of latent EBV infection in clinical samples49 (fig 1). However several recent studies suggest the existence of another form of latency lacking EBER expression.50,51
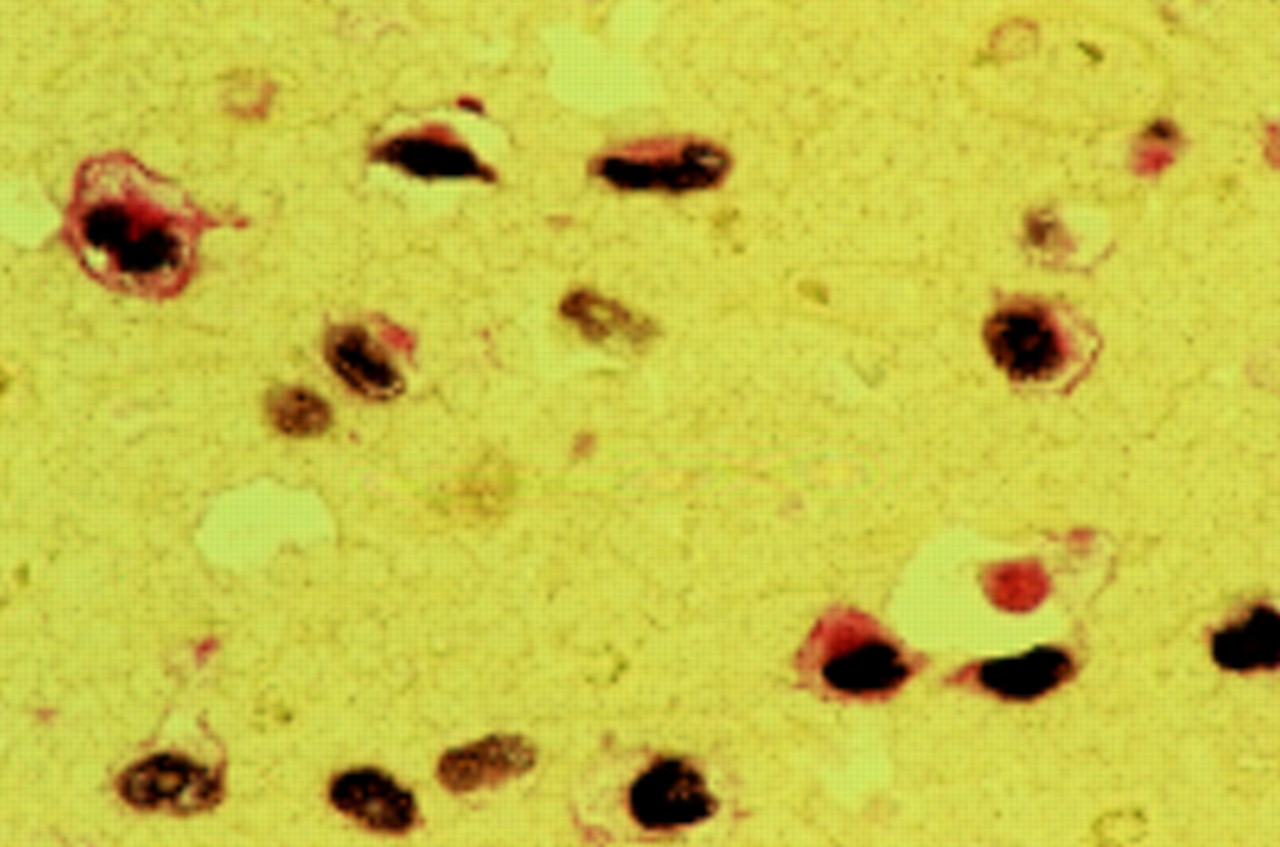
Double labelling of malignant Hodgkin-Reed Sternberg (HRS) cells showing co-expression of Epstein-Barr virus early RNAs (EBERs; brown/black) and latent membrane protein 1 (LMP1; red).
In Hodgkin's disease, the bulk of viral genomes are found in monoclonal form, indicating that infection of the tumour cells has occurred before their clonal expansion.47,52 EBV appears to persist throughout the course of Hodgkin's disease and is also found in multiple sites of Hodgkin's disease.53 Furthermore, the EBV genome copy number within HRS cells varies between patients but appears constant within individual patients with Hodgkin's disease.53 The inability to detect EBV in all cases of Hodgkin's disease could be the result of the failure to detect genomes present at a low copy number. However, this is unlikely, given the sensitivity and range of assays that have been used.54 Another possibility is that EBV transforms Hodgkin's disease progenitor cells by a “hit and run” mechanism,55 whereby EBV viral DNA rearrangement and loss occurs during malignant progression, similar to that recently suggested for some cases of sporadic Burkitt's lymphoma. However, Staratschek-Jox et al using fluorescence in situ hybridisation analysis found no evidence of integrated EBV genomes in EBV negative Hodgkin's disease tumours.56 Therefore, the most likely explanation is that EBV is associated with only a subset of patients.
The association of EBV with Hodgkin's disease seems to depend on factors such as country of residence, histological subtype, sex, ethnicity, and age. In particular, EBV positive Hodgkin's disease tumours appear to be less common in developed populations, with percentages of between 20% and 50% for North American and European cases,49,57–59 57% for Hodgkin's disease in China,60 but much higher rates in underdeveloped countries such as Peru61 and Kenya.62–64 The increased incidence of EBV positive Hodgkin's disease in underdeveloped countries could result from the existence of an underlying immunosuppression similar to that observed for African Burkitt's lymphoma in a malaria infected population.61 This is supported by the higher EBV positive rates in Hodgkin's disease from human immunodeficiency virus infected patients.65 Alternatively, the timing of EBV infection (which is likely to occur earlier in developing populations) might also be important.
EBV is more commonly associated with the MC subtype and less frequently with the other forms of this disease.54,66–68 In addition, Hodgkin's disease in the older age group and in children, especially boys under 10 years, has been shown to be more likely to be EBV associated than Hodgkin's disease in young adults.54,68–70 This suggested to Armstrong and colleagues70 that Hodgkin's disease consisted of three disease entities: Hodgkin's disease of childhood (EBV positive, MC type), Hodgkin's disease of young adults (EBV negative, NS type), and Hodgkin's disease of older adults (EBV positive, MC type). However, our data71 suggest a more homogeneous spread of EBV positive Hodgkin's disease within the adult age ranges defined by Armstrong. One likely explanation for these discrepancies is variation in the composition of Hodgkin's disease subtypes within each age group between studies. The infrequent association of EBV with Hodgkin's disease in young adulthood has also prompted the suggestion that a second virus might be involved, although there is little evidence to support this at present.72
Sex and ethnicity are also factors that are related to EBV positivity in Hodgkin's disease. Various studies have shown that EBV positive rates are higher in male patients than in female patients.68 In addition, international studies have indicated that EBV positive Hodgkin's disease affects more Asians (predominantly Chinese) and Hispanics than whites or blacks.68
Recently, we investigated socioeconomic factors by EBV status in Hodgkin's disease within a small regional population of the UK. Our results showed that higher levels of material deprivation, as determined by the Townsend score, were more likely in adult patients with EBV positive Hodgkin's disease compared with their EBV negative counterparts.73 This relation was particularly evident for female patients and for those with MC disease. Thus, it appears that socioeconomic differences might be responsible, at least in part, for some of the observed geographical variations in EBV positive rates in Hodgkin's disease.
HRS cells exhibit a type II form of latency, EBV gene expression being limited to the EBERs, Epstein-Barr nuclear antigen 1 (EBNA1),74 latent membrane protein 1 (LMP1)66,67 (fig 1), LMP2,75,76 and the Bam HIA transcripts.75 The particularly high level of LMP1 expression in HRS cells66,67 suggests that the virus is likely to be important in the pathogenesis of EBV associated cases. LMP1 induces many of the phenotypic changes seen in EBV infected B cells, including expression of the B cell activation markers, CD23 and CD40; IL-10 production; upregulation of cell adhesion molecules such as intercellular adhesion molecule 1 (ICAM-1), lymphocyte function associated antigen 1 (LFA1) and LFA3; and downregulation of CD99.77–79 LMP1 also protects B cells from cell death by the upregulation of several anti-apoptosis genes including bcl-2, mcl-1, and A20.80–82 LMP1 functions as a constitutively activated tumour necrosis factor (TNF) receptor and many of the phenotypic and growth transforming effects of LMP1 are the result of its ability to activate a variety of signalling pathways, including nuclear factor κB (NF-κB), through two C-terminus activating regions (CTAR1 and CTAR2).83–87
In fact, constitutive NF-κB activation has been consistently detected in HRS cells,88 and nuclear NF-κB expression can be observed in HRS cells by immunohistochemistry (P Murray, 1999, unpublished data) (fig 2). Inhibition of NF-κB activity in Hodgkin's disease cell lines leads to their increased sensitivity to apoptosis after growth factor withdrawal and their impaired tumorigenicity in severe combined immunodeficiency (SCID) mice.89 Although NF-κB activation is a common feature of HRS cells, the molecular routes to this activation may be different between EBV positive and EBV negative Hodgkin's disease. Thus, by single cell PCR of HRS cells, Jungnickel et al detected clonal mutations in the IκBα gene in two of three cases of EBV negative Hodgkin's disease, but no such defects in the two EBV positive cases examined.90 This suggests that the constitutive activation of NF-κB by LMP1 in EBV positive HRS cells may be substituted by IκBα gene mutations in HRS cells not infected by EBV.
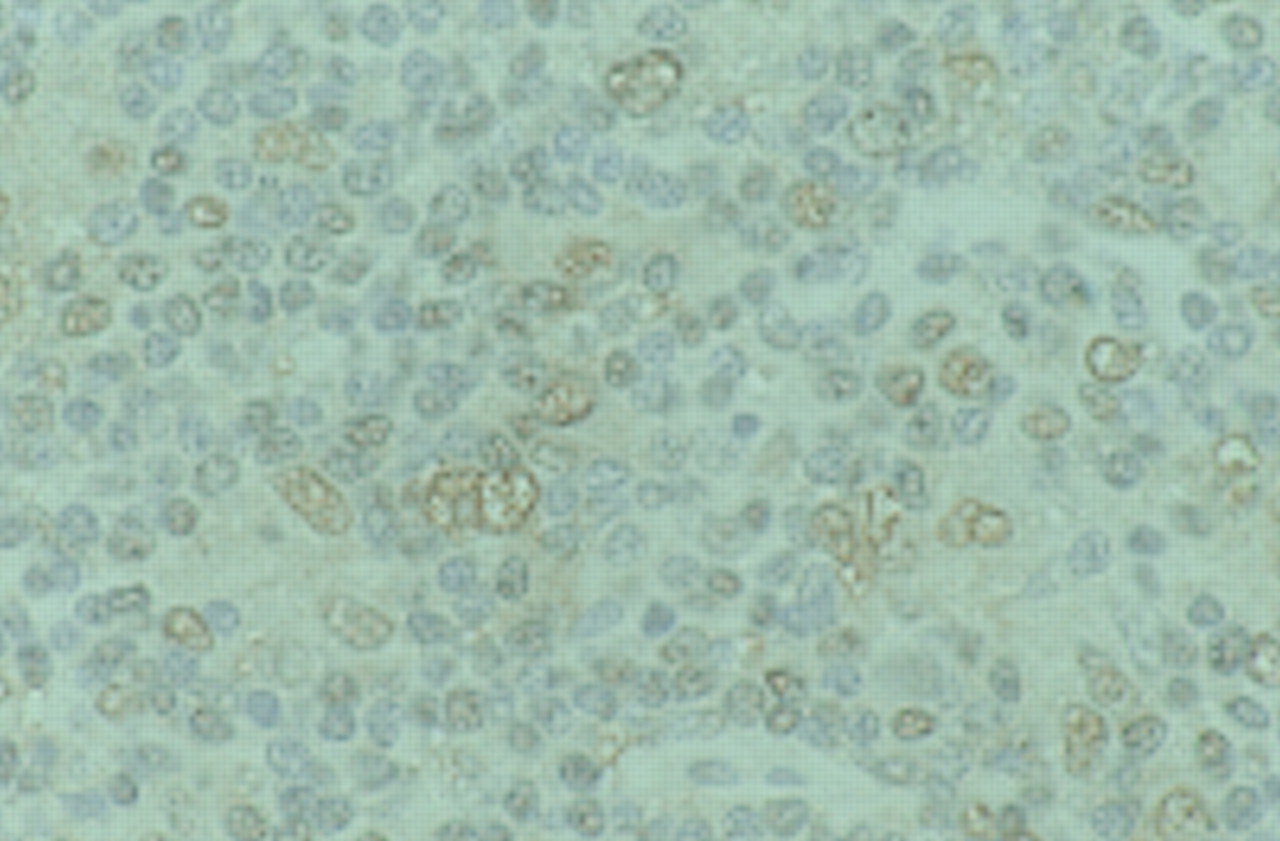
Immunohistochemical detection of nuclear factor κB (NF-κB) in the nuclei of Hodgkin-Reed Sternberg (HRS) cells and other lymphoid cells from Hodgkin's disease using an antibody that recognises the nuclear localisation signal of the p65 subunit of NF-κB.
Other studies on Hodgkin's disease have failed to show a correlation between LMP1 and the expression of many of the genes known to be upregulated by LMP1 in vitro. For example, BCL-2 protein concentrations do not correlate with LMP1 expression in Hodgkin's disease,91 but such a relation has been shown for post-transplant lymphomas.92 However, in other situations there is evidence that LMP1 regulated genes are more highly expressed in EBV positive, compared with EBV negative Hodgkin's disease, suggesting biologically important differences between the two. For example, IL-1093 and IL-694 are more frequently expressed in EBV positive compared with EBV negative Hodgkin's disease. Recent data show that TRAF1, which is upregulated by LMP1 in B cells in vitro is overexpressed in EBV positive Hodgkin's disease.95,96
A 30 bp deletion in the BNLF-1 gene, which encodes LMP1, in nude mouse propagated Chinese nasopharyngeal carcinoma (CAO) cells has been observed. CAO LMP1 was found to be more tumorigenic than the prototype B95.8 LMP1.97 Initially, this mutation was thought to be preferentially associated with nasopharyngeal carcinoma, but similar mutations have also been detected in some T cell lymphomas, Hodgkin's disease, infectious mononucleosis, and lymphoblastoid cell lines (LCLs) from healthy controls.98–100 Healthy virus carriers have been found to have a similar frequency of mutations to patients with virus infected tumours from the same geographical region.100 However, some studies have shown an increased incidence of this deletion variant in HIV positive Hodgkin's disease compared with HIV negative Hodgkin's disease,101 and also in paediatric Hodgkin's disease compared with normal controls.102
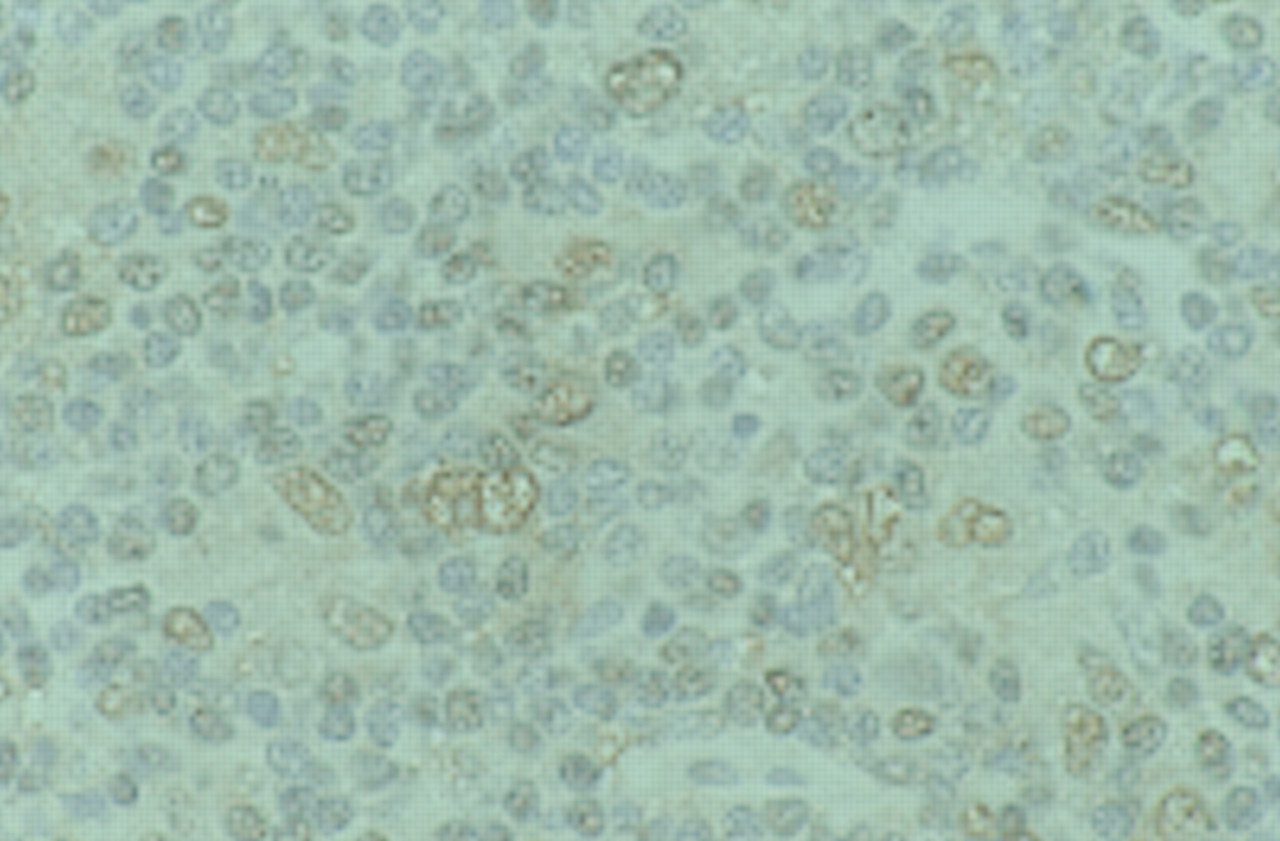
LMP2 is also expressed by HRS cells in EBV associated Hodgkin's disease (fig 3). LMP2A affects signal transduction by obstructing those pathways that are triggered by ligation of the B cell antigen receptor complex (see L S Young et al, this issue). Clustered plasma membrane patches of LMP2A and their N-terminal domains mimic crosslinked receptor tails and become phosphorylated on tyrosine and serine residues.103 The LMP2A molecules compete for the binding of the Src family protein tyrosine kinases and the Syk protein tyrosine kinases. This blocks signalling through the B cell antigen receptor complex and prevents transition of the EBV infected B cell into the lytic cycle and thus maintains EBV latency.104 The precise role of LMP2 in the pathogenesis of Hodgkin's disease, however, remains to be determined.

{kind=link}
{kind=link}
{kind=link}
Immunohistochemical detection of latent membrane protein 2 (LMP2) in Hodgkin's disease.
LMP2, and to a lesser extent LMP1, are targets for cytotoxic T cells in association with different major histocompatibility complex (MHC) class I restriction elements in vitro.105–108 The survival of EBV infected HRS cells in vivo suggests several possible explanations, including the existence of specific immunological defects present in patients with Hodgkin's disease that permit the growth of the neoplastic cells,52 or that EBV infected tumour cells have evolved strategies to evade immunosurveillance. Support for the latter is provided by the finding that IL-10 production is more frequent in EBV infected HRS cells when compared with their EBV negative counterparts, and this has been suggested to account for the failure of these cells to be recognised by EBV specific cytotoxic T lymphocytes (CTLs).93,109 This is further underlined by the observation that tumour derived T cells from EBV negative Hodgkin's disease show EBV specific cytotoxicity, whereas the corresponding cells from EBV positive Hodgkin's disease lesions do not.110 In fact, EBV positive cases of Hodgkin's disease have been shown to contain more activated CTLs and express relatively higher amounts of MHC class I molecules than EBV negative cases.108,111,112 Despite this, there is clearly a failure to elicit an effective anti-EBV CTL response.
One study has shown that the Hodgkin's disease cell line, HDLM2, is able to process and present epitopes from LMP1 and LMP2 in the context of multiple MHC class I alleles, including HLA A2, and is sensitive to lysis by EBV-specific CTLs.113 Furthermore, using autologous fibroblasts infected with a vaccinia recombinant encoding LMP2 as a target, the same authors were able to identify and expand LMP2 specific CTLs from the peripheral blood of a patient with Hodgkin's disease. The use of donor derived, EBV specific CTLs has also been investigated in the treatment of EBV positive patients with Hodgkin's disease.114 In this study, EBV specific CTLs could be generated from patients with advanced Hodgkin's disease, albeit at lower frequency than normal controls. EBV specific CTLs survived and had antiviral activity in vivo. These results provide some encouragement for the pursuit of CTL treatment for EBV associated Hodgkin's disease. However, further work is required to establish whether the microenvironment of EBV positive HRS cells is likely to compromise immunotherapeutic strategies targeted at EBV positive patients with Hodgkin's disease.
In this context, it is interesting that two large studies investigating the influence of EBV infection in Hodgkin's disease have shown improved outcomes for the EBV positive patients compared with their EBV negative counterparts.115,116 This is somewhat surprising when one considers that the oncogenic LMP1 protein is highly expressed in EBV infected HRS cells, but possible explanations could be that the malignant cells of EBV positive Hodgkin's disease are more sensitive to chemotherapy agents or that the EBV positive cells might be targets for immune cytolysis, particularly after cytoreduction by chemotherapy. The reported higher expression of bcl-2 in EBV negative Hodgkin's disease might suggest greater resistance to chemotherapy induced apoptosis in EBV negative tumours.91
Conclusion
Despite improvements in our understanding of the pathogenesis of Hodgkin's disease the precise contribution of EBV remains largely unknown. Future work to identify the roles of latent virus products, particularly LMP1 and LMP2, is therefore required. Such knowledge is likely to pave the way for greater refinement of EBV targeted gene therapies.