Article Text

Abstract
Aims: To determine whether there is an association between the insertion/deletion (I/D) polymorphism of the human angiotensin I converting enzyme (ACE) gene and malignant vascular injury (MVI).
Methods: The polymerase chain reaction was used to genotype DNA extracted from archival, paraffin wax embedded renal biopsy material from 48 patients with MVI, made up from cases of malignant hypertension (n = 23), scleroderma (n = 10), and haemolytic uraemic syndrome (n = 15), and from whole blood samples from 191 healthy controls.
Results: The D allele was found more frequently in cases of MVI than in healthy controls, (65% v 52%). Both the DD and I/D genotypes occurred significantly more frequently in patients with MVI than did the II genotype (χ2 = 7.26, p = 0.007; and χ2 = 4.06, p = 0.04, respectively).
Conclusions: Possession of at least one copy of the D allele is associated with an increased risk of developing MVI. Our data support a dominant mode of effect for the D allele. Use of the I/D polymorphism as a genetic marker for MVI may be of value clinically in identifying at risk individuals before the development of target end organ damage. Furthermore, those at risk may benefit from early ACE inhibition.
- end organ damage
- malignant hypertension
- angiotensin converting enzyme
- polymorphism
- ACE, angiotensin I converting enzyme; Ang, angiotensin; CI, confidence interval; EH, essential hypertension; HUS, haemolytic uraemic syndrome; I/D, insertional/deletional; MH, malignant hypertension; MVI, malignant vascular injury; OR, odds ratio; PCR, polymerase chain reaction; RAS, renin
- angiotensin system
Statistics from Altmetric.com
- ACE, angiotensin I converting enzyme; Ang, angiotensin; CI, confidence interval; EH, essential hypertension; HUS, haemolytic uraemic syndrome; I/D, insertional/deletional; MH, malignant hypertension; MVI, malignant vascular injury; OR, odds ratio; PCR, polymerase chain reaction; RAS, renin
- angiotensin system
Malignant vascular injury (MVI) is found in a heterogeneous group of relatively uncommon conditions, including malignant hypertension (MH), scleroderma, and haemolytic uraemic syndrome (HUS). Clinically, there is rapid and progressive renal impairment and circulatory collapse, which left untreated has a high mortality.1 The aetiologies of these conditions differ, but the unifying pathogenetic mechanism appears to be endothelial damage, followed by transmural vascular injury. This results in vascular wall remodelling with luminal narrowing, secondary renal ischaemia, and activation of the local renin–angiotensin system (RAS).2 The vascular lesions in MH, HUS, and scleroderma are similar histologically and are characterised by endothelial swelling, fibrinoid necrosis of afferent arterioles and terminal interlobular arteries, and concentric myointimal proliferation (endarteritis proliferans) of interlobular arteries.3,4 The clinical observation of an increased incidence of MH in certain ethnic groups supports a key role for genetic modifiers in determining susceptibility to MH.5 A recent study proposed the angiotensin I converting enzyme (ACE) gene as a candidate gene associated with the development of MVI in a rat model of MH (TGRmRen2–27), which carries the mouse Ren2 renin gene. A single homozygous male transgene donor was crossed with Fischer-Lewis F1 females. The offspring developed MH and early mortality. Genome wide screening and quantitative trait locus analysis showed linkage to chromosome 10, approximating to the locus of the rat ACE gene.6 Importantly, simultaneous studies showed that pharmacological ACE inhibition at non-hypotensive doses prevented the development of MH in susceptible TGRmRen2–27 animals.7
ACE is a zinc metallopeptidase that converts angiotensin I (Ang I) to angiotensin II (Ang II) and degrades bradykinin. It exists both as a membrane anchored form, on the surface of endothelial and epithelial cells, and also in a circulating plasma form.8 In 1990, Rigat et al characterised a 287 base pair (bp) insertion/deletion (I/D) polymorphism in intron 16 of the human ACE gene, which resides on chromosome 17q23,9 and has subsequently been associated with variance in both plasma and tissue concentrations of ACE, being highest in the DD genotype.10–13 Since then, the I/D polymorphism has been associated with an increased risk of either the development or the progression of several cardiovascular and renal diseases.14–17 Some studies have yielded conflicting results, including studies on essential hypertension (EH), where there have been both positive and negative reported associations.18–20 Most recently, two large prospective studies using different ethnic populations, the Japanese Suita and the American Framingham heart studies, have independently shown positive associations between the DD genotype and EH in men but not in women.21,22 Because there is considerable variability in terms of end organ damage between individuals with comparable blood pressures, increasing attention has focused on the contribution of genetic factors in determining target end organ damage in hypertension.23 Two recent Italian studies have shown an association between the D allele and target end organ damage in the kidney in EH.13,24 MH most commonly arises on a background of EH, it may also complicate secondary causes of hypertension such as renal parenchymal disease or renal artery stenosis, and it may also occur de novo. The trigger for progression to the malignant phase is poorly understood and can be observed over a range of blood pressures.25 As the most extreme form of hypertensive end organ damage, it may be of particular clinical importance to identify early and potentially treat those individuals with a genetic predisposition to the development of MH and other diseases characterised by MVI.
“The I/D polymorphism has been associated with an increased risk of either the development or the progression of several cardiovascular and renal diseases”
Our study was undertaken to establish whether there is an association between the I/D polymorphism of the ACE gene and the development of MVI in humans.
METHODS
Patients
A search was performed to identify patients from February 1987 to March 2000 who had undergone renal biopsy at Edinburgh Royal Infirmary, with a diagnosis of MH, scleroderma, or HUS. Paraffin wax blocks were retrieved from the archives of the department of pathology, University of Edinburgh for 51 patients. The median age was 53 years (range, 22–80) with a female to male ratio of 1.09. All subjects were white. Histology was reviewed by two pathologists (SF, NM) without knowledge of the clinical history. MVI was defined by the presence of at least two of the following: endarteritis proliferans of interlobular arteries, endothelial swelling, and/or fibrinoid necrosis of afferent arterioles. One hundred and ninety one control blood samples were obtained from the Scottish National Blood Transfusion Service. The anonymous blood donors were aged between 17 and 60 years and were healthy on routine examination. Ethical approval was obtained from the ethics committee, Lothian University Hospitals NHS Trust.
ACE genotype detection
Separate polymerase chain reaction (PCR) assays were used to amplify genomic DNA extracted from peripheral blood leucocytes (controls) and renal biopsy material (cases). Briefly, whole blood was washed in TE buffer (10mM Tris/HCl, 1mM EDTA, pH 8.0), centrifuged, and the supernatant removed. Leucocyte DNA was extracted using a lysis buffer containing 200 μg/ml of proteinase K, 50mM KCl, 10mM Tris, 2.5mM MgCl2, 0.1 mg/ml gelatine, 0.45% NP40, 0.45% Tween 20, and double distilled water. Samples were incubated at 50°C for 20 minutes, followed by boiling for 20 minutes. Kidney genomic DNA was extracted from formalin fixed, paraffin wax embedded tissue as described by Levi et al.26
The flanking oligonucleotide primer pairs used to characterise the I/D polymorphism anneal outside the 287 bp insertion within intron 16 of the ACE gene and produce a 490 bp fragment, corresponding to the I allele, and a 190 bp fragment, corresponding to the D allele and were as follows. Sense: oligo 5`- CTGGAGACCACTCCCATCCTTTCT-3`; antisense: oligo 5`-GATGTGGCCATCACATTCGTCAGAT-3`, as described previously.27 Owing to the preferential amplification of the D allele, mistyping of I/D heterozygotes as DD homozygotes is possible; therefore, insertion specific primer pairs were used to verify each DD genotype.28 These primer pairs produce a 335 bp PCR product only in the presence of the I allele and were as follows. Sense: oligo 5`-TGGGACCACAGCGC CCGCCACTAC-3`; antisense: oligo 5`-TCGCCAGCCCTCC CATGCCCATAA-3` (primers were supplied by GibcoBRL, Paisley, UK). Reaction mixtures consisted of 1.25 units of thermostable Taq polymerase (Advanced Biotechnologies, Epson, UK), 0.2mM of each dNTP, 1.5mM MgCl2, 2mM dimethyl sulphoxide (DMSO), and 100 ng of each primer, made up to a final volume of 50 μl. PCR assays were performed on an Omnigene Hybaid thermocycler (Ashford, UK). The cycle parameters to identify the I/D polymorphism from genomic DNA extracted from (1) control blood samples and (2) paraffin wax embedded renal biopsy material were as follows. (1) Thirty two cycles of 94°C for 30 seconds, 59°C for 30 seconds, and 72°C for one minute, followed by 59°C for one minute and 72°C for 10 minutes; (2) 40 cycles of 94°C for one minute, 58°C for one minute, and 72°C for two minutes, followed by 58°C for two minutes and 72°C for 10 minutes. For identification of the I allele cycling conditions were: (1) 32 cycles of 94°C for 30 seconds, 67°C for 30 seconds, and 72°C for 30 seconds, followed by 67°C for 30 seconds and 72°C for 10 minutes; and (2) 40 cycles of 94°C for one minute, 67°C for 45 seconds, 72°C for two minutes, followed by 67°C for 30 seconds and 72°C for 10 minutes. PCR products were analysed on a 1.5% agarose gel, stained with ethidium bromide, and visualised under ultraviolet irradiation.
Data analysis and power calculations
Associations between the I/D genotypes and allele frequencies in cases versus controls were analysed using the two tailed χ2 test. Required sample sizes were calculated using the statistical computer software Epi Info v6.04b, assuming a power of 80%, a significance level of 95%, a control to case ratio of 4 : 1, and an expected proportion of DD homozygotes in controls of 28%.29 A significant difference was defined as a two fold higher proportion of patients with the DD genotype in cases compared with controls. This produced minimum required sample sizes of 132 controls and 33 cases. An odds ratio (OR) and a 95% confidence interval (CI) were calculated to assess the relative risk conferred by each genotype and allele. A p value of < 0.05 was taken as significant.
RESULTS
Only a single patient failed to satisfy the histological criteria for the diagnosis of MVI and was excluded from further study. Figure 1 shows the histological features of MVI. Two cases failed to produce PCR amplification products with either the I/D or insert specific primers. The probable explanation for this was a combination of DNA degradation during storage (both biopsies were taken over 13 years ago) and the extremely small amount of tissue available. Forty eight cases produced PCR fragments of the expected sizes (fig 2). Thirteen of the 32 patients with MVI initially typed as DD homozygotes were reassigned to the I/D group after verification with the insert specific primer pairs. The overall frequencies of the II, I/D, and DD genotypes in the malignant vascular injury group were three (6%), 26 (54%), and 19 (40%), respectively, which were different to those obtained from the normal control group (40 (21%), 102 (53%), and 49 (26%), respectively; table 1). The allele frequencies were in equilibrium with that predicted by the Hardy-Weinberg equation (data not shown). The overall allele frequencies for the D allele were higher in the patients with MVI than in healthy controls; 65% and 52%, respectively (table 2). There was a significant increase in the frequencies of both the DD an I/D genotypes compared with II homozygotes in cases versus controls (χ2 = 7.26; p = 0.007; odds ratio, 5.17; 95% CI, 1.36 to 28.84 and χ2 = 4.06; p = 0.04; odds ratio, 3.4; 95% CI, 0.95 to 18.4, respectively), supporting a dominant mode of effect for the D allele.
Frequency of ACE I/D genotypes in cases of malignant vasular injury (MVI) and healthy controls
Overall frequency of ACE I/D alleles in cases of malignant vascular injury (MVI) and healthy controls
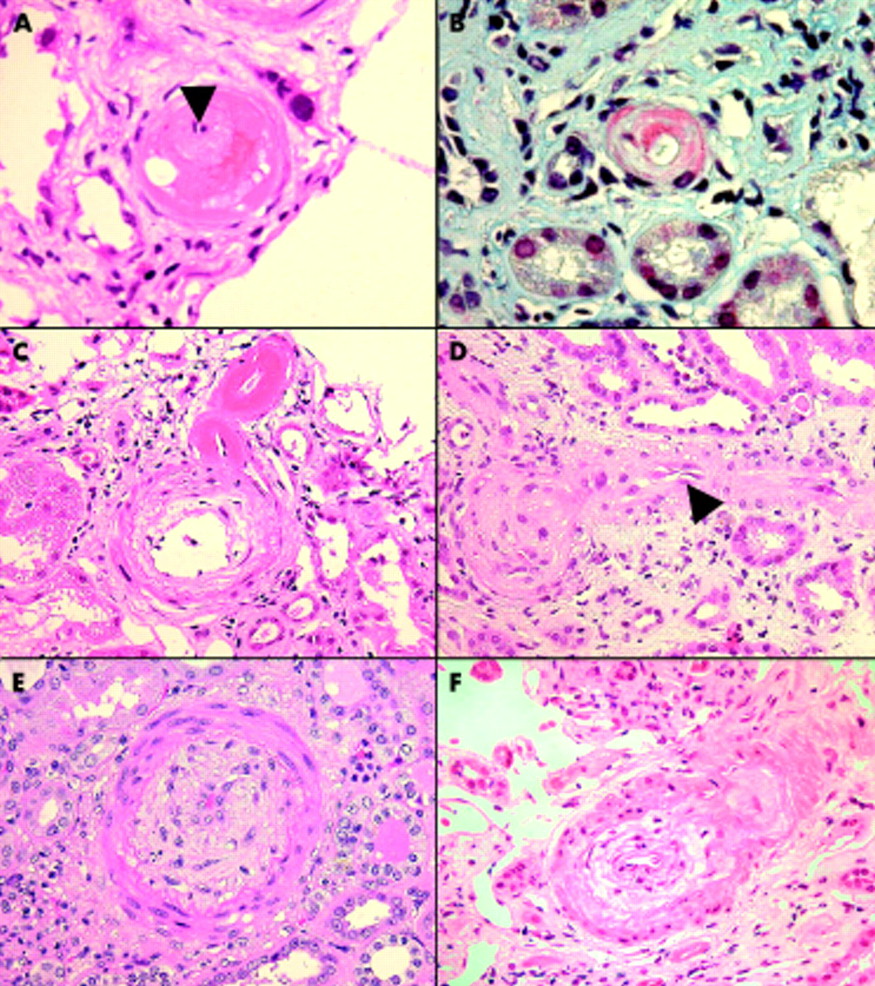
(A) Fibrinoid necrosis of a small interlobular artery. The vessel lumen is reduced to a pinpoint (arrow) and there is leakage of fibrin into the media (haematoxylin and eosin; original magnification, ×40). (B) Martius scarlet blue, staining fibrin within the wall of an afferent arteriole red (original magnification, ×40). (C) Concentric myointimal hyperplasia of an interlobular artery and fibrinoid necrosis of the afferent arteriole as it branches from the parent vessel (haematoxylin and eosin; original magnification, ×40). (D) Endothelial swelling (arrow), with almost complete obliteration of the lumen of an afferent arteriole (haematoxylin and eosin; original magnification, ×40). (E) Severe concentric myointimal hyperplasia “endarteritis proliferans” of an arcuate artery (haematoxylin and eosin; original magnification, ×40). (F) Mucoid oedema and concentric myointimal proliferation of an acutely injured interlobular artery (haematoxylin and eosin; original magnification, ×40).

{kind=link}
{kind=link}
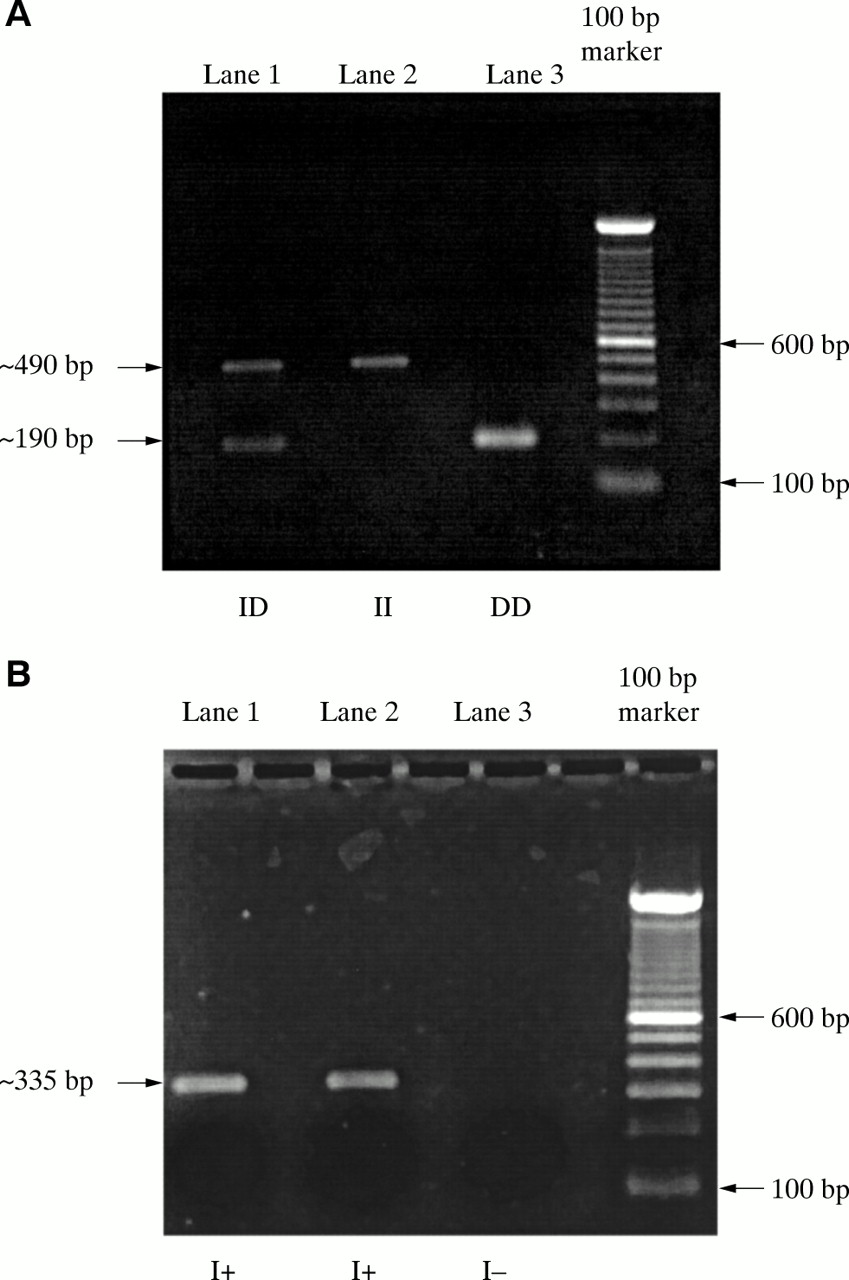
(A) Electrophoresis using flanking primer pairs produces the following PCR products: lane 1, I/D heterozygote with bands at both approximately 190 and 490 bp; lane 2, II homozygote with a single band of approximately 490 bp; lane 3, DD homozygote with single band at approximately 190 bp. (B) Electrophoresis using insert specific primers produces the following PCR products: lanes 1 and 2 both show single bands representing the insert fragment of 335 bp (I+), lane 3, no bands detected (I−), thus differentiating a true DD homozygote from an I/D heterozygote.
DISCUSSION
The ACE gene has recently been proposed as a candidate modifier locus for MH, following genetic linkage close to the ACE gene in a transgenic rat model of MH.6 An important role for activation of the local RAS in the pathophysiology of MVI is strengthened by the fact that pharmacological ACE inhibition using the same animal model prevented the development of the MH phenotype. This effect was observed independently of a reduction in systolic blood pressure, and was associated with decreased concentrations of tissue ACE, rather than circulating plasma ACE values.7 We designed our present study to investigate the association between the I/D polymorphism of the ACE gene and the risk of development of MVI in human subjects.
We have shown a positive association between the possession of the D allele and MVI in patients with MVI versus normal healthy control subjects. Furthermore, both the DD and I/D genotypes occurred significantly more frequently in patients with MVI than did the II genotype (patients: χ2 = 7.26; p = 0.007; controls: χ2 = 4.06; p = 0.04), suggesting that the possession of at least one D allele is associated with an increased risk of MVI. These results support the previous suggestion that the D allele exhibits a dominant mode of effect in determining target end organ damage.24 Our results are in agreement with Stefansson et al, who recently found a positive association between the DD genotype and the development of MH.30 This Swedish study diagnosed MH using the clinical criteria of severe hypertension (although the numerical blood pressure criteria used were not stated), associated with grade III or IV retinopathy. Renal biopsy material was only available in a small number of cases and no information was given regarding the presence of the recognised diagnostic features for MVI in any of these biopsies. The frequencies of DD genotypes in our patients with MVI were comparable: 40% compared with 43% in the Swedish study. However, their normal control subjects and non-malignant hypertensive groups both had DD genotype frequencies considerably lower than in previously reported European studies: 17.6% and 14.3%, respectively, versus 30%,13 39%,16 and 25%.23 In contrast, the DD frequency in our own healthy control population of 26% was close to that of a large meta-analysis including 135 studies and 19 065 subjects with a quoted frequency of 28%.29
“It remains possible that the D allele is a risk factor for development of scleroderma, rather than solely for malignant vascular injury”
Although more accurate in terms of classification, the use of renal biopsy material for histological diagnosis undoubtedly introduces potential sources of selection bias, because of variation in renal biopsy practice. Ultimately, the hospital renal biopsy population may not be truly representative of MVI in the general population. The anonymous nature of the blood donor samples used as normal controls made it impossible to age and sex match cases and controls, which is another source of possible bias. We chose a healthy control population, rather than hypertensives without end organ damage, because of the potential to include within this last group individuals with a genetic predisposition to develop MVI. The same rationale applies to the scleroderma group, whereby using a control population of scleroderma patients without MVI would inevitably include patients who will go on to develop renal involvement in the future. Indeed, scleroderma renal crisis usually occurs in patients with a history of scleroderma, which may be several years in duration.31 The true risk of developing MVI conferred by the D allele can only be established by prospectively following newly diagnosed patients with either scleroderma or hypertension, and subsequently identifying those who go on to develop MVI. It remains possible that the D allele is a risk factor for development of scleroderma, rather than solely for MVI.
Take home messages
-
The possession of at least one copy of the D allele is associated with an increased risk of developing malignant vascular injury (MVI) and the data support a dominant mode of effect for the D allele
-
The I/D polymorphism might be clinically useful as a genetic marker for MVI to identify at risk individuals before the development of target end organ damage
-
Those at risk may benefit from early angiotensin I converting enzyme inhibition
Owing to the nature of association studies, we cannot refute the possibility that the observed positive association we have found is the result of linkage disequilibrium between the ACE I/D polymorphism and another closely located functional polymorphic locus. However, there are several pieces of evidence supporting a functional role for the ACE I/D polymorphism and MVI. First, the DD genotype has been associated with increased serum and tissue concentrations of ACE.9,11 The ratio of Ang I to Ang II is higher in the kidney than in the serum, suggesting that the functional effect of the I/D polymorphism, in terms of local production of Ang II, may be of particular importance in the kidney.32 These effects are even more important following activation of the RAS, as occurs in MVI, and several recent reviews have highlighted the importance of the activation of the RAS in augmenting the effect of the DD genotype.33,34 Second, in animal models an MH phenotype can be induced by the infusion of Ang II. 35 Third, Ang II influences cell growth and matrix deposition, both of which are important in vascular wall remodelling.36,37 Indeed, there is an association of the D allele with myocardial infarction15 and with restenosis after coronary stent implantation,38 both of which reflect dynamic forms of vascular wall remodelling and involve activation of the local RAS.
Despite the lack of randomised controlled trials, there are data supporting the use of ACE inhibitors as first line drugs in the management of scleroderma renal crisis and MH.39,40 A recent prospective study has shown that the renoprotective effects, in terms of reduced proteinuria, were enhanced in individuals with the DD genotype.41 These findings, taken together with our own results, and the fact that in the TGRmRen2 rat model of MH pharmacological ACE inhibition at subhypotensive doses prevents the development of MH,7 leads us to propose that patients with the DD genotype, who appear to be at an increased risk of developing MVI, may benefit from early, prophylactic ACE inhibition.
In conclusion, we have shown a significant association between the D allele of the ACE gene and MVI. In the future, large scale prospective clinical trials are needed to determine whether similar associations are reproducible for the ACE gene and other components of the RAS, across different ethnic populations. The possible protective effects of ACE inhibition against the development of MVI should also be further investigated.
Acknowledgments
This study was jointly funded from a Pathological Society of Great Britain and Ireland clinical trainee grant and a National Kidney Research Fund grant.